Combining X-ray Crystallography and Solution NMR Data
X-ray crystallography and NMR Spectroscopy are two leading analytic techniques for elucidation of molecular structure at atomic resolution. Over than 99% of all deposited protein structures are resolved by either X-ray crystallography or NMR spectroscopy. X-ray crystallography provides precise determination of a unique macromolecular structure, whereas NMR spectroscopy is able to detect dynamics in solution, thus, these two approaches are highly complementary, and integration of two techniques gives valuable information on protein structure determination.
Structural restraints that can be obtained in NMR spectroscopy, such as pseudo-contact shifts (PCSs) and residual dipolar coupling (RDCs) have proved very useful for solving protein structure. Recently, PCSs and RDCs have been used for structure refinement in combination with X-ray data, by implementing them as structural restraints in the program REFMAC5. REFMAC5 is an extension of the crystallographic refinement program of REFMAC-NMR. REFMAC-NMR is used for medium resolution structure, by the inclusion of PCS and RDC NMR data as structural restraints in addition to the X-ray diffraction pattern. It takes advantage of the structural information contained in both the available crystallographic model and in the primary X-ray data. It performs a structural refinement by the simultaneous use of X-ray and NMR data (figure 1), and can be used to detect the presence of significant discrepancies, outside the uncertainty related to the experimental dataset, between solution and crystal structures.
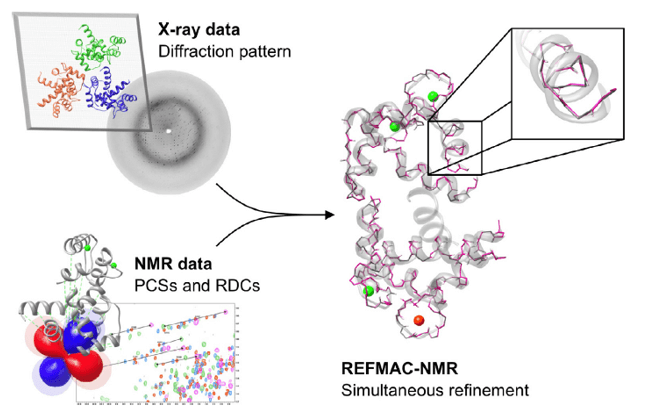 Figure 1. Information used in REFMAC-NMR structure refinement
Figure 1. Information used in REFMAC-NMR structure refinement
The general approach used by REFMAC-NMR for the inclusion of NMR data in the structure refinement consists of two minimizations. The first minimization against the X-ray data alone; the second minimization performed with the same weight setting including the NMR data (i.e. PCSs, RDCs), in order to decrease the discrepancy between experimental and back-calculated data. When all domains composing a system have been satisfactorily refined against X-ray and NMR data, the best-fit tensors obtained for each domain could be compared.
This integrated approach based on the combination of data from these two radically different sources yields structures with improved quality, and allows for a deeper understanding of the behavior of the biomolecules in solution.
The Protein Crystallography platform and NMR platform at Creative Biostructure are supported by a group of experts specialized in NMR and X-ray Crystallography. We are happy to help our customers to solve protein structures by incorporates both solid and solution state data from X-ray Crystallography and NMR, respectively. Please feel free to contact us through online inquiry.
Ordering Process

Reference
- Carlon A., Ravera E., Andralojc W., Parigi G., Murshudov G., Luchinat C. (2016) How to tackle protein structural data from solution and solid state: An integrated approach. Progress in Nuclear Magnetic Resonance Spectroscopy. 92-93: 54-70.

