Dynamic Conformational Change and Function Identification
Visualization of the interaction between a ligand and its protein target is of great importance to understand the exact molecular mechanisms and the associated physiological processes in cells. Despite the powerfulness of X-ray crystallography as a means to show interactions between atoms within complex molecules, it only provides data on a stationary basis such that the conformational change and dynamic patterns of the tested molecule are largely ignored. Cryo-electron microscopy (Cryo-EM), on the other hand, has undergone a quantum leap progression in resolution and becomes an essential technique in structural biology for dynamic studies of functional complexes not only in their physiological state, but also in their natural environment in the cell. Therefore, Cryo-EM can provide unique information on the conformational flexibility of macromolecular complexes, which are often unstable, flexible, scarce or transient in their native environments. The studying in model dynamics and conformational transitions allows for identification of the function of macromolecular complexes in cell.
Currently, there are two methods used in studying the conformational variability analysis from cryo-EM images: discrete-state methods and continuous-state methods. Discrete-state methods may provide a limited view of data heterogeneity because that only a small number of classes was used, whereas continuous-state methods offer a broader view of data heterogeneity than discrete-state methods, leading to an increased accuracy of the dynamic studies of macromolecular complexes. There are some noteworthy examples that have recently been published, for example, understanding the conformational changes is necessary for the function of ATPase p97 (Figure 1). The Cryo-EM structures give an insight into nucleotide-driven structural changes that drive function and show how inhibitor binding prevents these conformational changes.
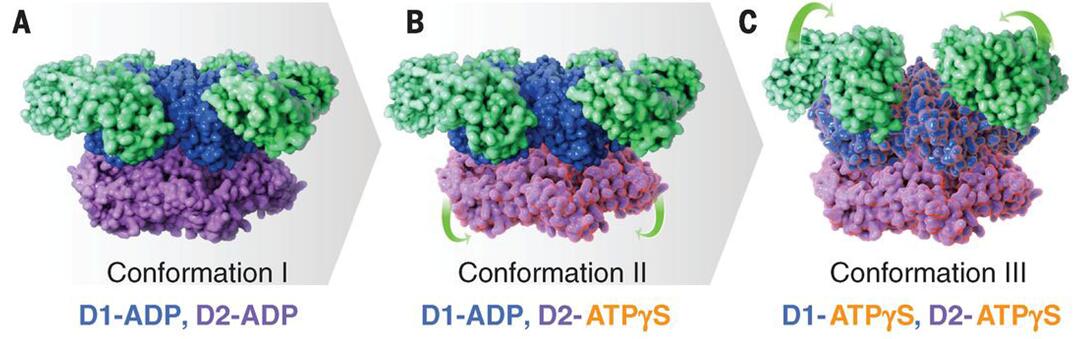 Figure 1. Cryo-EM structures at ~3.3 Å, ~3.2 Å, and ~3.3 Å resolution, respectively, of three distinct p97 conformational states populated upon addition of ATPγS.
Figure 1. Cryo-EM structures at ~3.3 Å, ~3.2 Å, and ~3.3 Å resolution, respectively, of three distinct p97 conformational states populated upon addition of ATPγS.
Creative Biostructure offers cutting-edge instruments and workstations to enable rapid and efficient Cryo-EM analysis. Our technical team consists of seasoned scientists with rich experiences in structural biology and Cryo-EM operation. Creative Biostructure promises to work closely with our customers to meet their specific needs and requirements.
Please feel free to contact us for a detailed quote.
Ordering Process

References
- Catherine Ve´nien-Bryan, Zhuolun Li, et al. Cryo-electron microscopy and X-ray crystallography: complementary approaches to structural biology and drug discovery. Acta Crystallogr F Struct Biol Commun. 2017;73:174-183.
- Soojay Banerjee, Alberto Bartesaghi, et al. 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science. 2016; 351: 871-875.
- SlavicaJonić, Computational methods for analyzing conformational variability of macromolecular complexes from cryo-electron microscopy images. Curr Opin Struct Biol. 2017; 43: 114-121.

