Importance of Protein Structure Determination
Understanding protein structure reveals its functional behavior. Living systems depend on proteins as essential molecules whose three-dimensional arrangements control their activities and interactions with other molecules. The structures of proteins determine how they interact with their ligands or substrates through specific binding mechanisms and conformations of functional regions. Despite its historic status as the top method for protein analysis traditional X-ray crystallography struggles to work with many proteins that resist crystallization. Cryo-EM technology analyzes protein structures directly through non-crystallized samples which makes it effective for studying macromolecular complexes and membrane proteins.
Cryo-EM technology helps researchers explore better ways to design drugs based on protein structures. Researchers study protein and protein complex structures at high resolution to learn about disease processes and make better drugs. Cryo-EM technology helps researchers study protein structure changes in their natural state.
Key Advantages of Cryo-EM in Protein Structure Analysis
The key advantages of cryo-EM in protein structure analysis include:
- No need for protein crystallization: Cryo-EM can directly analyze uncrystallized protein samples, solving the problem of difficult protein crystallization in traditional techniques.
- High-resolution capability: In recent years, the resolution of Cryo-EM has reached the atomic level (close to 1 Å), which can analyze complex biological macromolecular structures, such as membrane proteins and macromolecular complexes.
- Native state analysis: Cryo-EM can analyze proteins under close to physiological conditions, providing information on their dynamic conformation and functional state.
- Flexibility and diversity: Cryo-EM is suitable for a variety of sample types, including small molecule complexes, membrane proteins, and macromolecular complexes.
- Support for drug development: The high-resolution structures analyzed by Cryo-EM provide important support for structure-based drug design, especially in new drug discovery and vaccine development.
- Data integration and computational modeling: Combining deep learning and computational modeling techniques, Cryo-EM data can be used to predict protein interactions, dynamic properties, and functional regions.
How Cryo-EM Works
1. Sample Preparation
Sample preparation is a crucial step in Cryo-EM experiments, which directly affects the data quality and the resolution of the final results. The following are the key steps of sample preparation:
- Sample preparation: Samples usually include biomacromolecules such as proteins, viruses, and organelles. First, the target sample needs to be purified by biochemical methods and ensure its homogeneity (such as a single conformation or minimal conformational heterogeneity).
- Sample gridding: The sample solution is coated on a carbon film and covered with a thin layer of ice to form an ice shell. This step requires rapid cooling of the sample to prevent ice crystals from forming, thereby maintaining the sample in a near-native state.
- Sample evaluation: Before high-resolution data acquisition, a preliminary evaluation of the sample is required, such as diagnostic Cryo-EM imaging to check whether the sample is suitable for subsequent analysis.
2. Data Collection
Data collection is the process of acquiring a large number of two-dimensional projection images through a transmission electron microscope (TEM). The following are the main steps of data collection:
- Electron beam irradiation: A high-energy electron beam is used to irradiate the sample from multiple angles to generate a series of two-dimensional projection images. These images are called "micrographs".
- Image acquisition: Tomography forms a series of tilt series (a tilt series is a collection of images taken from different angles) by rotating the sample and taking images at each angle. These images are then transferred to a computer for further processing.
- Data quality control: During data acquisition, the exposure time and electron dose need to be monitored to ensure that the image quality meets the requirements of subsequent analysis.
3. Image Processing and 3D Reconstruction
Image processing and 3D reconstruction are key steps in converting 2D projection images into 3D structural models. The following are the main processing steps:
- Motion correction and dose weighting: First, the micrograph is motion corrected to eliminate image blur caused by sample motion. Then the image is weighted according to the electron dose to balance the signal intensity in different areas.
- Particle picking: Particles of interest are selected from the original micrograph and segmented. This step can be done manually or with automated tools.
- 2D classification: The selected particles are classified according to similarity to reduce the complexity of subsequent calculations.
- 3D reconstruction: The 2D projection images are reassembled into a 3D model through an iterative algorithm. Commonly used algorithms include Fourier transform-based methods and recently developed deep learning methods.
- Model refinement: The preliminary reconstructed 3D model is optimized and refined to improve resolution and accuracy.
Select Service
Cryo-EM in Soluble Protein Structure Determination
Cryo-EM has made breakthrough progress in recent years and has been able to resolve structures with near-atomic resolution. For example, in 2020, scientists successfully resolved the structure of human heme-binding proteins (such as Apoferritin) using single-particle cryo-EM technology, reaching a resolution of 1.25 Å. This level of resolution is sufficient to clearly observe the position of individual atoms, providing unprecedented details for understanding protein catalytic mechanisms and drug-protein interactions.
Case Study: Apoferritin Cryo-EM Structure
Apoferritin is an iron storage protein that has become an important model protein for cryo-EM studies due to its stable structure and easy access.
Analysis Challenges
The main challenges faced in the structural analysis of apoferritin include:
- Difficulty in sample preparation: Since apoferritin needs to remain stable at low temperatures, the sample preparation process needs to be specially optimized.
- Resolution Limitation: Although cryo-EM technology has been able to achieve a resolution of 1.25 Å, further improving the resolution still requires overcoming technical bottlenecks such as large data volume and complex calculations.
- Density map quality: High-quality density maps are the key to achieving high-resolution analysis, but how to optimize the quality of density maps remains a challenge.
Solution
- Optimize sample preparation: Sample quality can be improved by improving the sample preparation process, such as using specific cryogens (such as propane) to avoid ice crystal formation.
- Improve computing power: Optimize particle selection and image processing processes using GPU acceleration and deep learning algorithms (such as SPHRe-CryoEM), significantly improving data processing efficiency.
- Hardware upgrade: The application of new electron microscopes (such as Titan Mono-BCOR) and high-resolution imaging equipment makes it possible to resolve high-resolution structures.
Analysis Results
Using the above method, scientists successfully analyzed the 1.25 Å resolution structure of Apoferritin and achieved the visualization of single atoms for the first time. This achievement not only provides important information for understanding protein function, but also opens up new paths for drug design and disease treatment.
 Figure 1. High-Resolution Cryo-EM Reconstruction of Apoferritin. (Nakane T, et al., 2020)
Figure 1. High-Resolution Cryo-EM Reconstruction of Apoferritin. (Nakane T, et al., 2020)
Cryo-EM Applications for Membrane Proteins
Membrane proteins are difficult to crystallize due to their natural lipophilicity and easy aggregation in aqueous solution, so traditional X-ray crystallography methods are difficult to resolve their structures. However, Cryo-EM technology provides a new research approach for membrane proteins, which can directly resolve the three-dimensional structure of membrane proteins under near-physiological conditions.
Use of Nanodiscs for Membrane Protein Stabilization
Nanodiscs are a ring-shaped structure formed by a lipid bilayer encapsulating a protein molecule, which is widely used in the stabilization and structural analysis of membrane proteins. Studies have shown that Nanodiscs can effectively maintain the native conformation of membrane proteins and reduce the complexity of sample preparation. For example, using Nanodiscs, researchers have successfully analyzed the high-resolution structures of a variety of membrane proteins, including G protein-coupled receptors (GPCRs) and ion channels.
The application of Nanodiscs not only improves the stability of membrane proteins, but also solves the problem of easy inactivation of membrane proteins in aqueous solutions in traditional methods. In addition, by combining other auxiliary tools (such as detergents or specific ligands), Nanodiscs further enhance the analytical ability of membrane proteins.
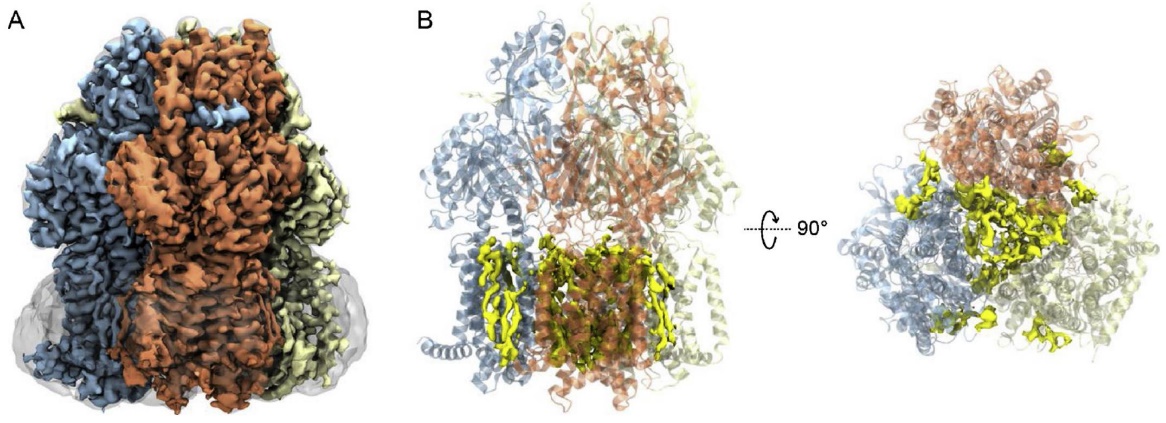 Figure 2. Cryo-EM Structure of AcrB in SMA Nanodiscs. (A) 3D reconstruction of the AcrB trimer with each protomer uniquely colored. The SMA nanodisc is shown as a smoothed transparent surface. (B) Cartoon representation of the AcrB structure with EM density of lipids (yellow) visualized in the central cavity and annular sites. (Sun C, et al., 2019)
Figure 2. Cryo-EM Structure of AcrB in SMA Nanodiscs. (A) 3D reconstruction of the AcrB trimer with each protomer uniquely colored. The SMA nanodisc is shown as a smoothed transparent surface. (B) Cartoon representation of the AcrB structure with EM density of lipids (yellow) visualized in the central cavity and annular sites. (Sun C, et al., 2019)
Select Service/Product
Related Reading
Application of Membrane Proteins Embedded in Liposomes
Liposomes are a technology that simulates the cell membrane environment and can effectively stabilize membrane proteins and maintain their functional activity. Studies have shown that after embedding membrane proteins in liposomes, Cryo-EM can be used to analyze their structure and study their interactions with ligands or other molecules. This method not only improves the stability of membrane proteins, but also provides a new perspective for studying their dynamic processes.
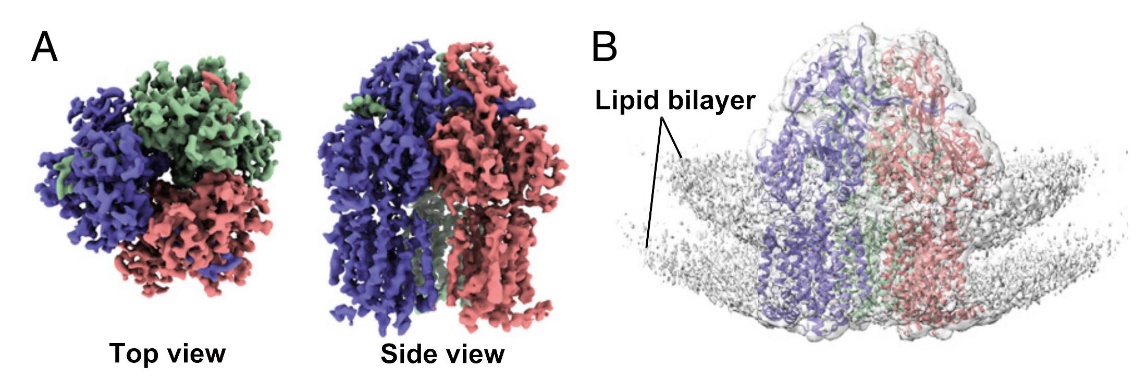 Figure 3. Single-Particle Cryo-EM Reconstruction of AcrB in Proteoliposomes. (A) Top and side views of AcrB reconstruction at 3.9 Å resolution. (B) EM map showing lipid bilayer signals with the AcrB structure docked at 1.0σ density contour. (Yao X, et al., 2020)
Figure 3. Single-Particle Cryo-EM Reconstruction of AcrB in Proteoliposomes. (A) Top and side views of AcrB reconstruction at 3.9 Å resolution. (B) EM map showing lipid bilayer signals with the AcrB structure docked at 1.0σ density contour. (Yao X, et al., 2020)
Select Service/Product
Related Reading
Progress in GPCR Structural Analysis
GPCRs are important targets for drug development, but their small size and the characteristics of being embedded in lipid membranes make Cryo-EM analysis challenging. However, in recent years, multiple GPCR structures have been successfully analyzed by using technologies such as Nanodiscs and nanobodies. For example, using Nanodiscs combined with detergents or ligands, researchers have revealed the conformational changes of GPCRs in different states, thus providing new directions for drug design.
Nevertheless, for some small molecular weight (such as 35-45 kDa) GPCR complexes, Cryo-EM analysis still faces certain limitations. However, with the advancement of technology, such as the introduction of voltage phase plates, the application of Cryo-EM in GPCR research is expected to be further expanded in the future.
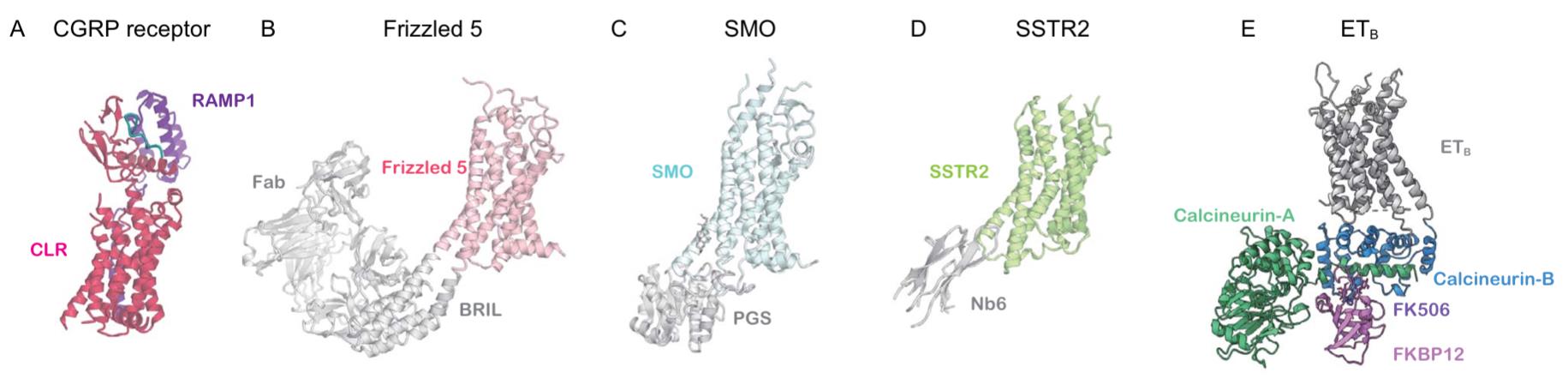 Figure 4. Cryo-EM structures of inactive GPCRs: (A) CGRP receptor bound to peptide (7KNU); (B) Frizzled 5-BRIL with antibodies (6WW2); (C) SMO-PGS fusion (8CXO); (D) SSTR2-Nb6 (7UL5); (E) ETB-calcineurin fusion (8JK9). (Shihoya W, et al., 2024)
Figure 4. Cryo-EM structures of inactive GPCRs: (A) CGRP receptor bound to peptide (7KNU); (B) Frizzled 5-BRIL with antibodies (6WW2); (C) SMO-PGS fusion (8CXO); (D) SSTR2-Nb6 (7UL5); (E) ETB-calcineurin fusion (8JK9). (Shihoya W, et al., 2024)
Select Service
Related Reading
- Structural Research of G Protein-coupled Receptors (GPCRs) Class A
- Structural Research of G Protein-coupled Receptors (GPCRs) Class B1
- Structural Research of G Protein-coupled Receptors (GPCRs) Class B2
- Structural Research of G Protein-coupled Receptors (GPCRs) Class C
- Structural Research of G Protein-coupled Receptors (GPCRs) Class D
- Structural Research of G Protein-coupled Receptors (GPCRs) Class F
Cryo-EM in Small Protein Structure Determination
Challenges in Small Protein Structure Determination
1. Low signal-to-noise ratio
Due to their small size, small molecular weight proteins (<50 kDa) have low sample contrast and low signal-to-noise ratio (S/N ratio), which makes image reconstruction difficult. For example, many small molecular weight proteins lack unique low-frequency structural features, making it difficult to achieve accurate single-particle image alignment. In addition, the offset of particles in ice crystal samples and signal-to-noise ratio issues further limit the resolution of small molecular weight proteins.
2. Difficulty in sample expression and purification
Small molecular weight proteins are usually low in expression, highly heterogeneous, and poorly stable, especially in the presence of detergents, all of which affect the quality of Cryo-EM imaging. In addition, small molecular weight proteins are prone to conformational changes under dynamic conditions, which increases the difficulty of structural resolution.
3. Resolution limitation
Although Cryo-EM technology has made significant progress in recent years, the resolution of small molecular weight proteins is generally still limited to 2-4 Å, and higher resolution (such as atomic level) is still difficult to achieve. For example, some studies have found that even for smaller proteins such as hemoglobin αβ complex, the resolution can only reach about 3 Å.
4. Technical bottlenecks
The main bottlenecks faced by current Cryo-EM technology in analyzing small molecular weight proteins include limitations in sample preparation, imaging hardware, and image processing algorithms. For example, traditional methods have difficulty capturing atomic-level details of small molecular weight proteins, while high-resolution atomic models require higher S/N ratios and more precise image alignment techniques.
Recent Advances and Case Studies
In recent years, scientists have developed a variety of new tools and methods to overcome the limitations of small molecular weight protein structure determination. For example, rigid imaging supports have been used to improve the imaging quality of small molecular weight proteins. In addition, nano-antibody extended single particle Cryo-EM technology (Megabodies) has also been shown to significantly improve the resolution of small molecular weight proteins.
1. High-resolution cases
Although the Cryo-EM resolution of most small molecular weight proteins is still below 3 Å, some successful cases have shown that higher resolution can be achieved by optimizing sample preparation and imaging conditions. For example, the Cryo-EM structures of small molecular weight proteins such as lactate dehydrogenase and isocitrate dehydrogenase have been successfully resolved to 3.8 Å or even higher.
2. Advances in computational methods
Computational methods have played an important role in the analysis of Cryo-EM data. For example, deep learning technology has been used to improve the reconstruction quality and analysis accuracy of Cryo-EM images. In addition, combined with computational methods such as molecular dynamics simulation, the analysis of Cryo-EM data can be further optimized.
3. Combining experimental and computational methods
Combining experimental and computational methods has become an important strategy to solve the problem of small molecular weight protein structure determination. For example, by combining Cryo-EM data and X-ray crystallography data, the accuracy of structure elucidation can be improved.
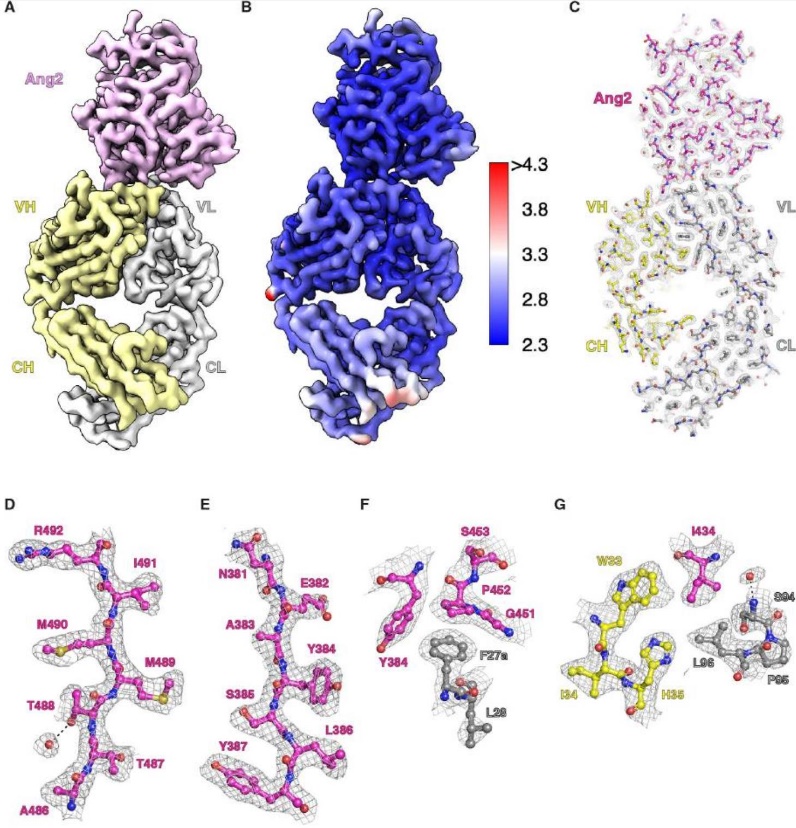 Figure 5. Cryo-EM structure of the Ang2-5A12.6DS Fab complex at 2.7 Å resolution. (A) Overall structure with Ang2 in pink, Fab HC in yellow, and LC in light gray. (B) Local resolution map of the complex. (C-G) EM density for the entire complex (C) and selected high-resolution regions (D-G), highlighting Ang2 (magenta), RTX LC (gray), and HC (yellow). (Kung J E, et al., 2024)
Figure 5. Cryo-EM structure of the Ang2-5A12.6DS Fab complex at 2.7 Å resolution. (A) Overall structure with Ang2 in pink, Fab HC in yellow, and LC in light gray. (B) Local resolution map of the complex. (C-G) EM density for the entire complex (C) and selected high-resolution regions (D-G), highlighting Ang2 (magenta), RTX LC (gray), and HC (yellow). (Kung J E, et al., 2024)
Select Service
Cryo-EM in Dynamic Protein Complex Structural Analysis
Challenges in the Structural Analysis of Dynamic Protein Complexes
1. Low signal-to-noise ratio
Although cryo-EM technology can provide three-dimensional structural information of protein complexes, its image quality is limited by the signal-to-noise ratio, which makes it difficult to conduct high-throughput experiments and obtain a large number of high-resolution structures.
2. Difficulty in capturing dynamic processes
The dynamic properties of protein complexes make it complicated to capture transient states in a single frozen sample. In addition, the analysis of dynamic processes requires the combination of other techniques (such as NMR or molecular dynamics simulation) to verify and supplement cryo-EM data.
3. Difficulty in analyzing small molecules
For smaller proteins or complexes, cryo-EM has limited ability to analyze their atomic-level structures because the resolution is usually less than 3 Å, while atomic-level details require higher resolution.
4. Heterogeneity and complexity
There may be significant conformational heterogeneity within protein complexes, which places higher requirements on data processing and model building.
Recent Technological Advances and Case Studies
1. Time-resolved cryo-EM
Time-resolved cryo-EM is an advanced technology for studying the dynamic processes of protein complexes. It can capture the structural changes of proteins in different states, thereby revealing their functional mechanisms. Time-resolved cryo-EM captures transient intermediates of protein complexes by stopping biochemical reactions at specific time points and combining them with rapid freezing techniques (such as vitrification). This method is particularly suitable for studying short-lived structural changes, such as the activation process of GPCRs.
2. Application of deep learning and artificial intelligence
Deep learning technology plays an important role in Cryo-EM data processing. For example, algorithms such as AlphaFold2 significantly improve the efficiency and accuracy of protein structure analysis by predicting the three-dimensional structure of amino acid sequences. In addition, Transformer-based generative models are also exploring the conformational space of protein complexes.
3. Combining with other technologies
In order to overcome the limitations of Cryo-EM in resolving dynamic processes, researchers have combined technologies such as NMR, mass spectrometry (MS), and molecular dynamics simulations. For example, the method combining Cryo-EM and cross-linked mass spectrometry (CX-MS) has been used to resolve macromolecular complexes.
Select Service
- Cryo-EM for Protein Complex
- Dynamic Conformational Change and Function Identification
- NMR Spectroscopy Services
- Molecular Dynamics Simulations
- Integrative Structural Biology Services
- Model Reconstruction of Crystal Structures Using Cryo-EM and NMR
- Combinational Applications of Cryo-EM, X-ray Crystallography and NMR
- Combinational Applications of Cryo-EM and Mass Spectrometry
In summary, Cryo-EM has transformed the landscape of protein structure determination, offering unparalleled advantages in the analysis of both soluble and membrane proteins. Recent advances in small protein and dynamic protein complex analysis illustrate Cryo-EM's versatility in tackling some of the most challenging aspects of structural biology.
At Creative Biostructure, we offer cryo-EM services for protein structure analysis, and support drug discovery projects with a focus on target protein structures. Our expertise in Cryo-EM can help drive your research forward, whether you are investigating new protein targets or designing structure-based therapeutics. If you are looking to leverage Cryo-EM for your next project, don't hesitate to contact us for customized solutions and advanced structural analysis.
References
- Cheng Y. Membrane protein structural biology in the era of single particle cryo-EM. Current opinion in structural biology. 2018, 52: 58-63.
- Sun C, Gennis R B. Single-particle cryo-EM studies of transmembrane proteins in SMA copolymer nanodiscs. Chemistry and physics of lipids. 2019, 221: 114-119.
- Nakane T, Kotecha A, Sente A, et al. Single-particle cryo-EM at atomic resolution. Nature. 2020, 587(7832): 152-156.
- Yao X, Fan X, Yan N. Cryo-EM analysis of a membrane protein embedded in the liposome. Proceedings of the National Academy of Sciences. 2020, 117(31): 18497-18503.
- Klebl D P, Aspinall L, Muench S P. Time resolved applications for Cryo-EM; approaches, challenges and future directions. Current Opinion in Structural Biology. 2023, 83: 102696.
- Shihoya W, Iwama A, Sano F K, et al. Cryo-EM advances in GPCR structure determination. The Journal of Biochemistry. 2024, 176(1): 1-10.
- Kung J E, Johnson M C, Jao C C, et al. Disulfi de constrained Fabs overcome target size limitation for high-resolution single-particle cryo-EM. bioRxiv, 2024.
- Papasergi-Scott M M, Pérez-Hernández G, Batebi H, et al. Time-resolved cryo-EM of G-protein activation by a GPCR. Nature. 2024, 629(8014): 1182-1191.

-1.jpg)