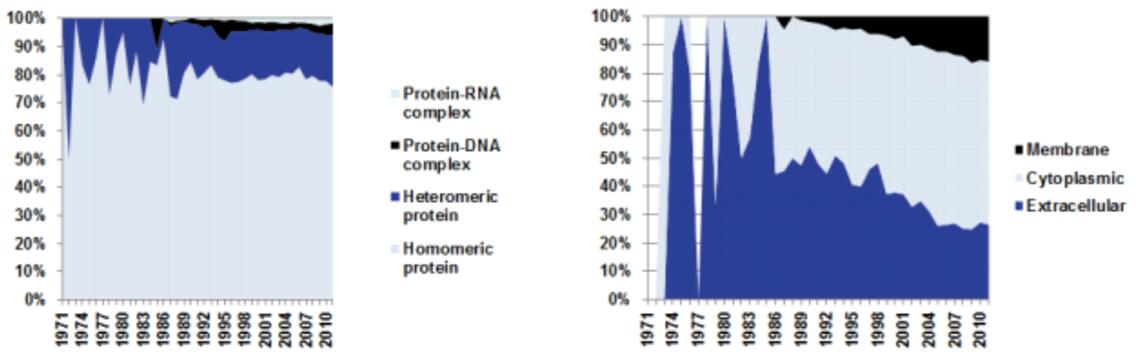
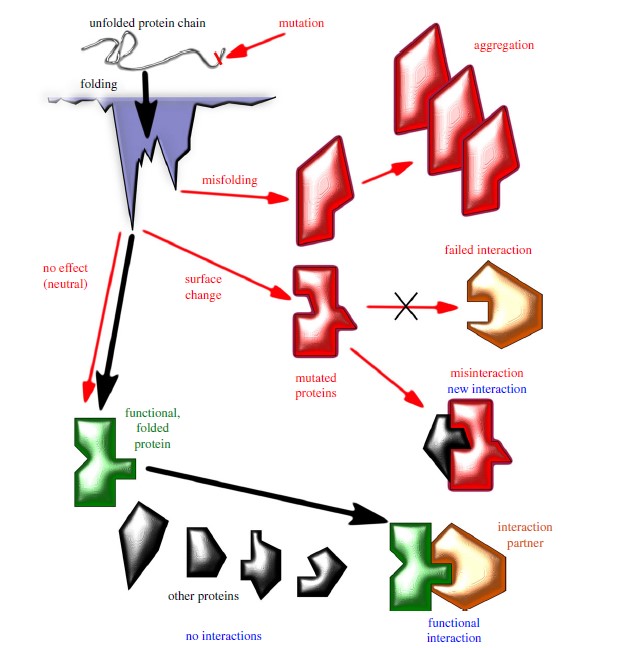
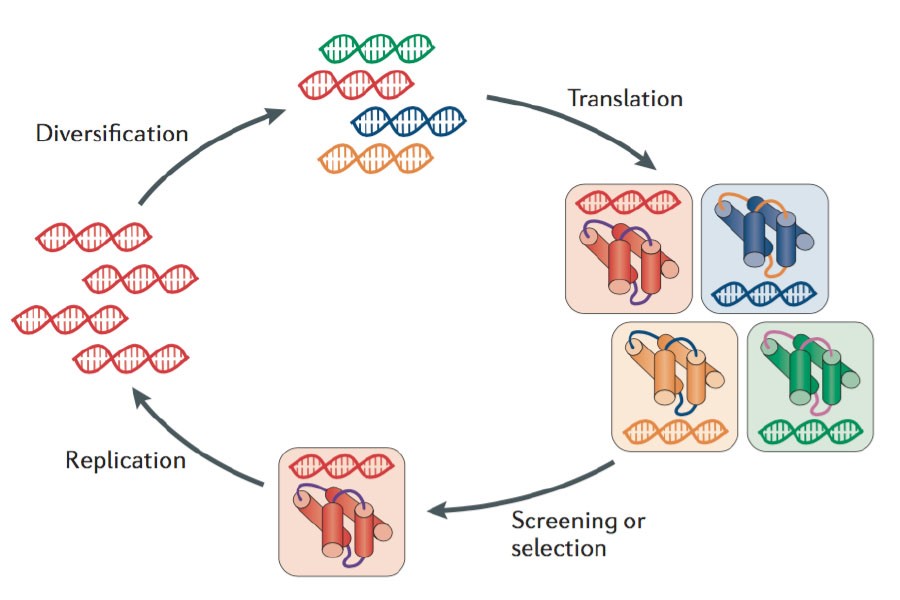
AlphaFold represents a groundbreaking advancement in the field of structural biology and protein structure prediction. For decades, scientists have been grappling with the challenge of predicting the 3D structure of proteins from their amino acid sequences, a problem essential for understanding biological processes, drug discovery, and disease mechanisms. AlphaFold's evolution, from its first version to its third, has brought transformative insights and revolutionized the field.
What is AlphaFold 1?
Building upon previous machine learning techniques and utilizing neural networks, AlphaFold 1 was trained on protein structures from the Protein Data Bank (PDB). At the heart of this effort was addressing the protein-folding problem, a longstanding challenge that had perplexed the scientific community for over 50 years.
At the 13th Critical Assessment of Protein Structure Prediction (CASP13) in 2018, AlphaFold 1 made its debut and demonstrated remarkable results. It achieved the highest accuracy ever seen in the competition, outperforming conventional methods and setting a new benchmark in protein structure prediction.
Key Innovations of AlphaFold 1
AlphaFold 1 employed multiple sequence alignments (MSA) and incorporated evolutionary information. By using neural networks to predict the backbone torsion angles and residue-residue distances, it introduced a novel energy minimization strategy based on gradient descent, thereby predicting the 3D conformation of proteins.
Comparison with Previous Methods
Compared to earlier techniques that relied on homology modeling or physics-based simulations, AlphaFold 1 introduced machine learning into the prediction pipeline, which significantly improved the accuracy and speed of predictions. However, it was still constrained by limitations in handling complex protein structures or large datasets.
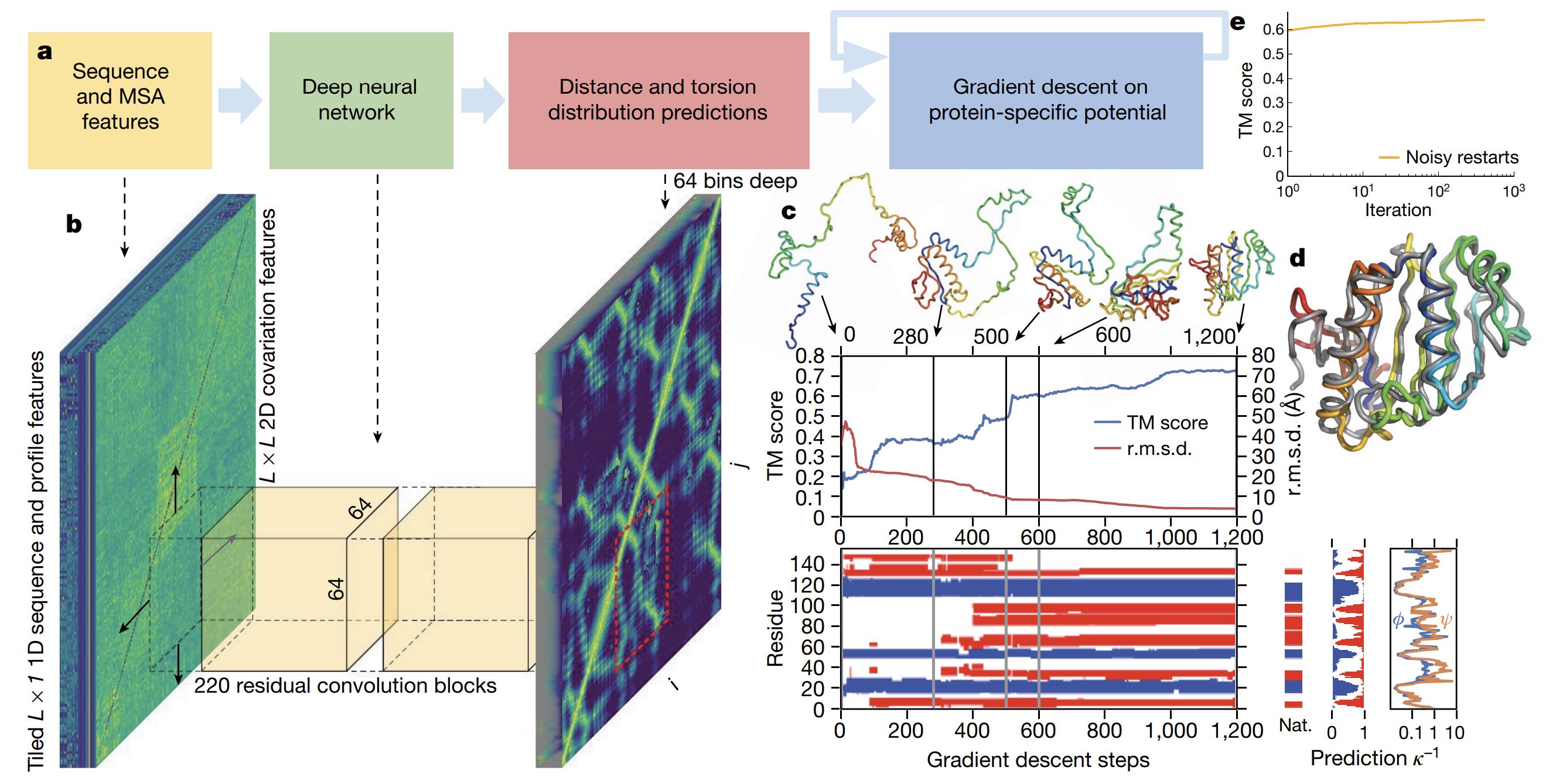 Figure 1. The folding process illustrated for CASP13 target T0986s2 using AlphaFold 1. (Senior A W, et al., 2020)
Figure 1. The folding process illustrated for CASP13 target T0986s2 using AlphaFold 1. (Senior A W, et al., 2020)
What is AlphaFold 2?
In 2020, DeepMind introduced the second generation of AlphaFold (AlphaFold 2) at the 14th Critical Assessment of Protein Structure Prediction (CASP14) competition, where it stunned the scientific community with unprecedented accuracy. AlphaFold 2 secured first place with remarkable all-atom Root Mean Square Deviation (RMSD) precision of 1.5 Å and backbone accuracy of 0.96 Å, creating a vast gap between it and the second-place competitor (which achieved an all-atom RMSD of 3.5 Å and backbone accuracy of 2.8 Å). To put this into perspective, a carbon atom's diameter is roughly 1.4 Å, demonstrating the extraordinary precision of AlphaFold 2's predictions.
Technical and Methodological Enhancements
Unlike AlphaFold 1, AlphaFold 2 is a completely redesigned neural network model, integrating deep learning algorithms with evolutionary, physical, and biological characteristics of protein structures. Its architecture fuses multi-sequence alignment (MSA) data and pairwise residue relationships. By introducing the Evoformer module, AlphaFold 2 could better handle evolutionary data and refine predictions. This was complemented by the Structure module, which iteratively refined the model outputs to achieve unparalleled precision.
Neural Network Architecture
AlphaFold 2's architecture was completely revamped to enable end-to-end protein structure prediction. It utilized a deep neural network trained on a vast dataset of protein sequences and structures. The neural network embedded the MSA and structural information simultaneously, enabling accurate predictions of both backbone geometry and side-chain positions.
Data Utilization and Training
The dataset used for training AlphaFold 2 included hundreds of thousands of protein sequences, along with high-quality structures from the PDB. The model was designed to generalize from this data, predicting novel protein structures with minimal input beyond the amino acid sequence.
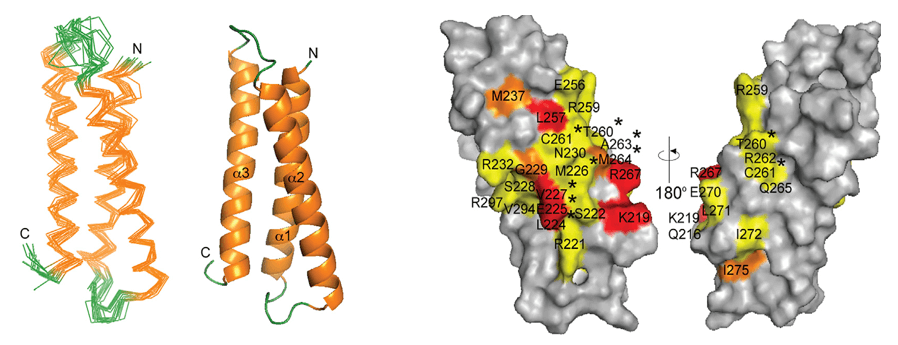
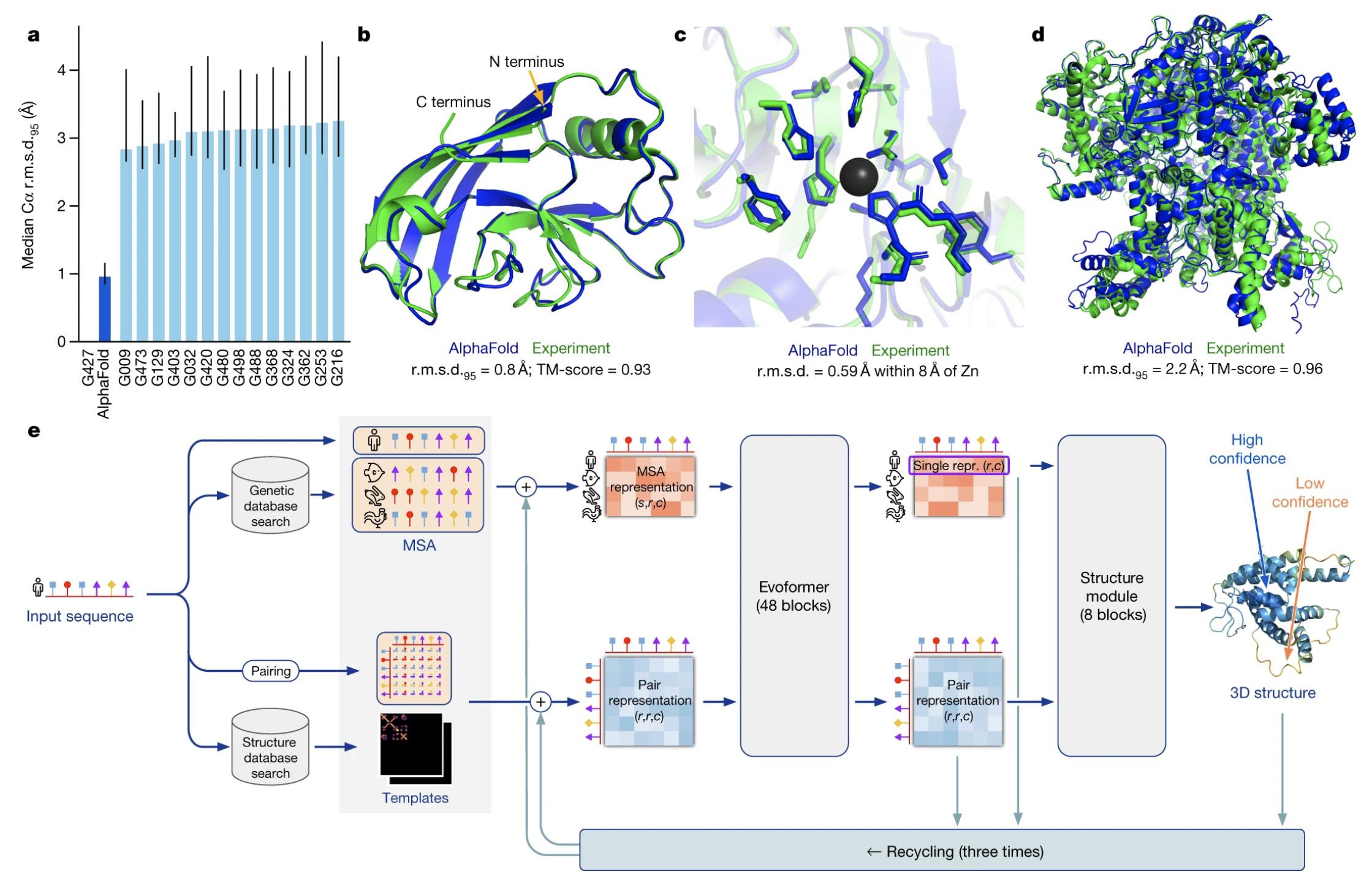 Figure 2. Highly accurate structures produced by AlphaFold 2. (Jumper J, et al., 2021)
Figure 2. Highly accurate structures produced by AlphaFold 2. (Jumper J, et al., 2021)
Transformative Impact of AlphaFold 2 on Protein Structure Prediction
AlphaFold 2's ability to predict protein structures with near-experimental accuracy has been recognized as a historic achievement in computational biology. This breakthrough has opened new avenues for understanding complex biological mechanisms, studying diseases, and accelerating drug development.
- Revolutionizing Structural Biology: The influence of AlphaFold 2 on structural biology has been profound. It enables researchers to predict protein structures rapidly, significantly reducing the time and cost associated with experimental methods like X-ray crystallography and cryo-electron microscopy. The structural biology community has embraced AlphaFold 2 as an indispensable tool for deciphering the intricate 3D conformations of proteins.
- Accelerating Research and Drug Discovery: Previously, determining the structure of a protein could take months or even years of labor-intensive experimental work. Now, with AlphaFold 2, the same task can be completed in hours using standard CPUs. Numerous recent research projects and drug discovery efforts have relied on AlphaFold 2's structural predictions, showcasing its transformative impact.
However, despite its groundbreaking achievements, AlphaFold 2 does have limitations. Its predictions are restricted to single protein structures, and it cannot predict the interactions between proteins and other molecules, such as multi-subunit protein complexes, DNA, or RNA. Additionally, it struggles with predicting molecular interactions. These limitations have since been addressed with the release of AlphaFold 3, which improves prediction accuracy and expands its applicability to a wider range of biomolecules.
What is AlphaFold 3?
AlphaFold 3 represents a major leap forward in protein structure prediction, expanding on AlphaFold 2's architecture while introducing significant innovations in both scope and precision.
Key Improvements Over AlphaFold 2
- Expanded Scope: AlphaFold 3 breaks new ground by accurately predicting not only protein structures but also the complexes involving DNA, RNA, antibodies, small molecules, ions, and covalent modifications. This capability marks a significant departure from AlphaFold 2, which was primarily focused on single protein structures. The new model's predictions align closely with experimentally determined structures, far outperforming other popular software tools like AutoDock Vina and RoseTTAFold.
-
Architectural Changes:
- Pairformer Module: One of the most notable changes in AlphaFold 3 is the replacement of AlphaFold 2's Evoformer module with the simpler Pairformer module. This update reduces the amount of multi-sequence alignment (MSA) data that needs to be processed, thereby improving computational efficiency without sacrificing accuracy.
- Diffusion Module: Another key innovation is the introduction of the Diffusion module, which predicts the raw atomic coordinates of the protein structure. This replaces the Structure module, which in AlphaFold 2 handled the specific torsion angles of amino acid backbones and side chains. The Diffusion module eliminates many of the complex operations that were previously part of the Structure module, such as bond-specific adjustments, while retaining or even enhancing prediction accuracy.
- Enhanced Prediction Accuracy: AlphaFold 3 has made revolutionary improvements in prediction accuracy, particularly for protein-DNA and protein-antibody interactions, achieving accuracy rates that are double those of AlphaFold 2. In addition, it outperforms conventional methods for drug interaction predictions. For instance, in benchmarking tests like PoseBusters, AlphaFold 3 demonstrated a 50% improvement over the best traditional methods, which significantly enhances its utility in pharmaceutical and biotechnological applications.
- Improved Computational Efficiency: By streamlining the neural network architecture and simplifying the complexity of structure handling, AlphaFold 3 operates with much higher computational efficiency. It achieves faster predictions with fewer resources, making it more accessible for large-scale projects and high-throughput structure predictions.
- Broad Applicability: AlphaFold 3 serves as a unified tool for predicting virtually all forms of biomolecular interactions. It is versatile enough to handle diverse biological macromolecules, including proteins, nucleic acids, antibodies, small molecules, ions, and post-translational modifications. This universal capability underscores its status as a revolutionary tool in structural biology.
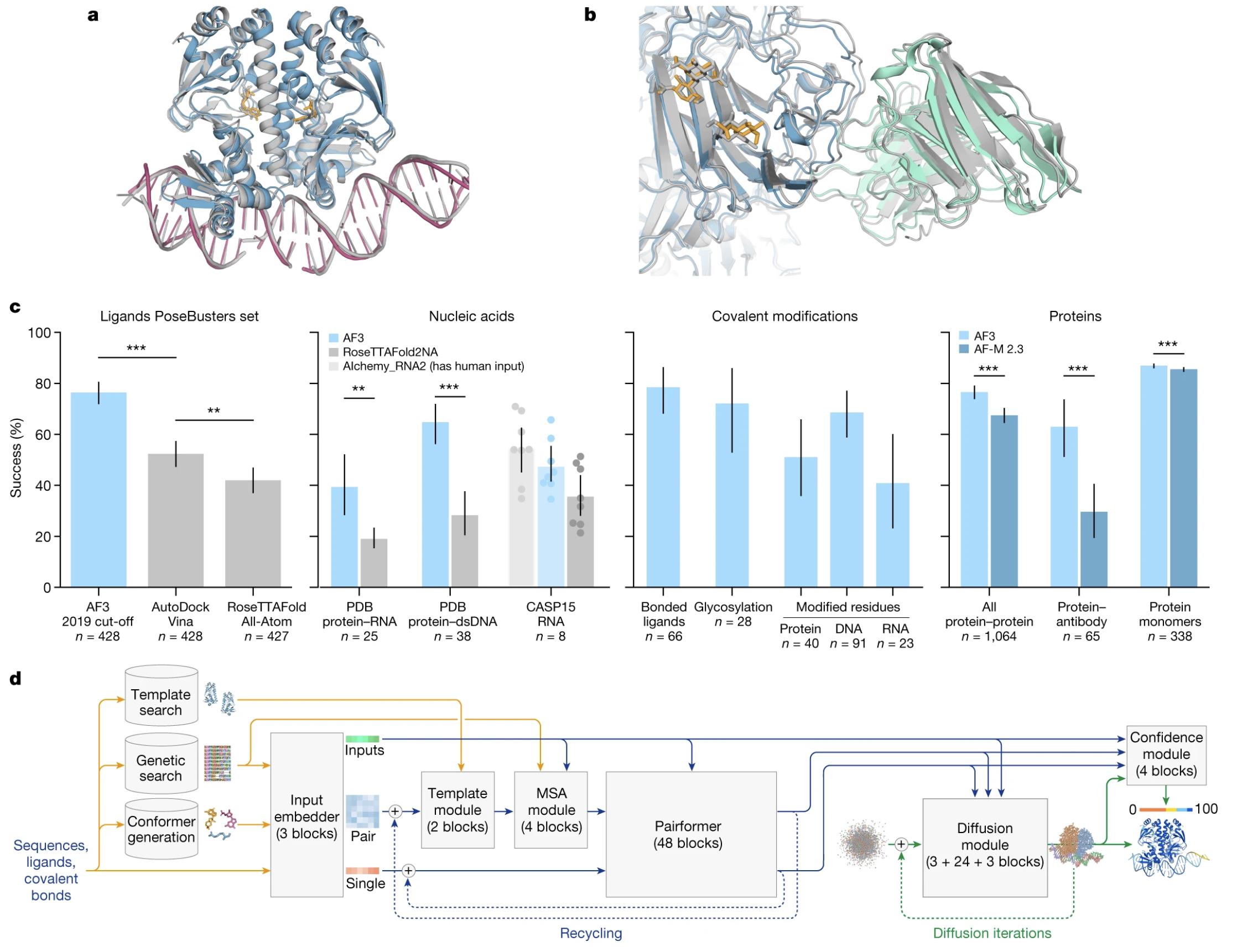 Figure 3. Structures across biomolecular complexes are accurately predicted by AlphaFold 3. (Abramson J, et al., 2024)
Figure 3. Structures across biomolecular complexes are accurately predicted by AlphaFold 3. (Abramson J, et al., 2024)
Limitations of AlphaFold 3
Despite its groundbreaking advances, AlphaFold 3 still faces several challenges:
- Stereochemistry: The predicted models occasionally exhibit stereochemical issues, including chirality errors (~4.4%) and atomic clashes due to overlapping atoms.
- Hallucination Effect: In regions of disorder, AlphaFold 3 may generate false structures, known as the hallucination effect. Although these regions are often flagged as low-confidence predictions, they are less distinguishable from valid predictions than in AlphaFold 2.
- Dynamics: Like its predecessors, AlphaFold 3 produces static structures and does not account for the dynamic behavior of molecules in solution, which remains a key limitation in protein structure prediction.
- Accuracy for Certain Targets: AlphaFold 3 may produce incorrect or incomplete conformations for specific proteins. For example, in predicting the structure of E3 ubiquitin ligase, it consistently produces a closed conformation, even when the protein should adopt an open conformation in the absence of a ligand.
To address these limitations, AlphaFold 3 incorporates several strategies, such as penalizing chirality errors and atomic clashes during model training and refining hallucination effects using cross-distillation methods. While these techniques reduce errors, they do not completely eliminate them. Generating multiple predictions and ranking them can improve accuracy, though at the cost of increased computational demands.
Competitive Landscape and Complementary Tools
While AlphaFold has garnered most of the attention, several other models have contributed to the protein-folding field. Tools such as RoseTTAFold, developed by the Baker Lab, and AutoDock Vina, known for ligand docking, have provided complementary capabilities. These tools are valuable for different types of predictions, especially in cases where AlphaFold's accuracy may be limited.
The success of AlphaFold has spurred collaborative efforts across the scientific community. The AlphaFold Protein Structure Database, which offers over 200 million predicted protein structures, has enabled unprecedented access to structural data. This open-access initiative has fostered collaboration in areas ranging from human health to environmental research.
While AlphaFold 3 represents a major breakthrough, there are still challenges to address. Future iterations could further refine stereochemistry, dynamic behavior, and complex protein-ligand interactions. Enhanced training data, including more diverse structural examples, could lead to even more accurate predictions.
Creative Biostructure focuses on integrating computational tools and experimental methods for comprehensive protein structure analysis. By collaborating across disciplines, we aid clients in incorporating AlphaFold predictions into their research, thereby expediting advancements in structural biology and drug development. The structural insights from AlphaFold 3 enable precise protein modifications, enhancing both efficiency and success rates to meet diverse protein requirements.
Our services compensate for AlphaFold 3's predictive limitations by combining experimental data with computational predictions to produce accurate structural models. We offer dynamic conformation analysis, capturing protein conformations in various environments, including interactions with ligands. Additionally, we provide guidance for protein engineering and drug discovery, using AlphaFold 3 predictions to accelerate drug design through experimental verification and optimization. For further details about our services and solutions, don't hesitate to contact us.
References
- Senior A W, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020. 577(7792): 706-710.
- Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021. 596(7873): 583-589.
- Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024: 1-3.