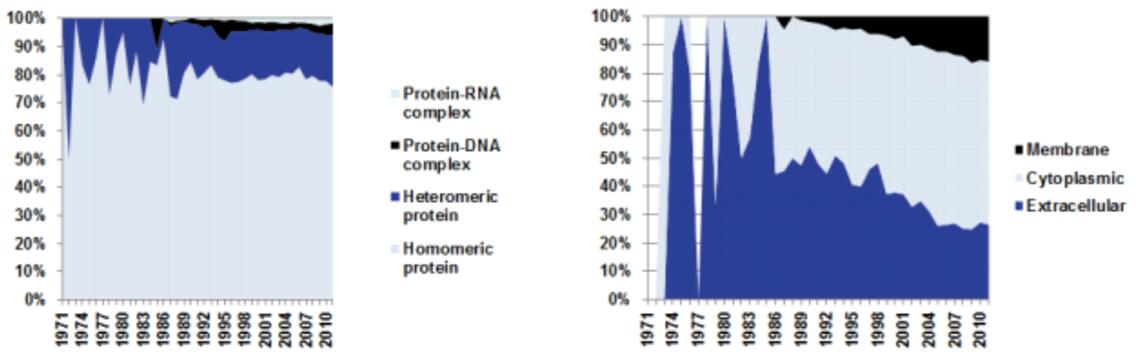
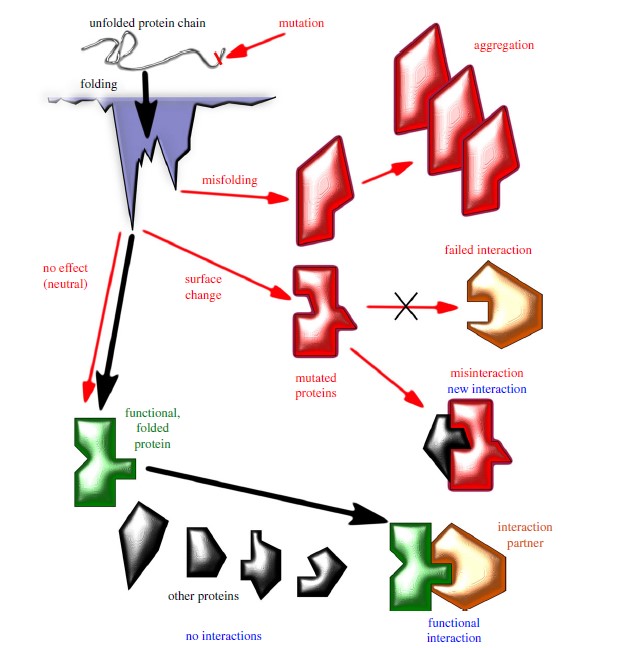
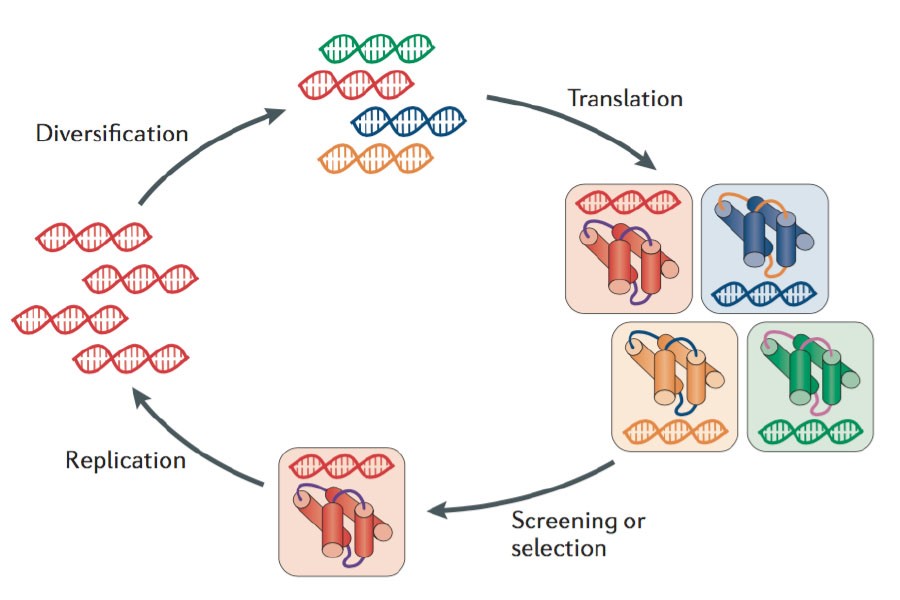
AlphaFold3 (AF3) is the latest version of the AlphaFold series, aimed at predicting molecular interactions with high accuracy. AlphaFold2 (AF2) had already revolutionized protein structure prediction, and AF3 extends those capabilities to a much broader scope, including protein-ligand, protein-nucleic acid, and other complex interactions. The AF3 model surpasses previous iterations in its ability to predict the structures of various biomolecules, including small molecules, nucleic acids, proteins, and covalently modified residues.
This development opens new frontiers in drug discovery, protein engineering, and molecular biology research, allowing scientists to model protein-ligand docking, nucleic acid interactions, and other complex molecular configurations with unmatched accuracy.
AlphaFold3 Model Architecture
AF3 achieves its exceptional results through significant improvements to the AF2 architecture, followed by extensive training. The goal was to expand AF3's ability to process a broader range of molecular data while improving learning efficiency. Key architectural enhancements include:
- Pairformer Module: AF3 replaces AF2's Evoformer module with a simpler Pairformer. This reduces the amount of Multiple Sequence Alignment (MSA) processing, making the system faster and more efficient while maintaining high accuracy. The Pairformer is responsible for modeling the relationships between pairs of molecules, handling various interaction types.
- Diffusion Module: AF3 uses a diffusion-based model to predict raw atomic coordinates, replacing AF2's Iterative Point Attention (IPA) structure module. The diffusion process operates across multiple scales—low noise helps the network improve local structure, while higher noise levels enable better prediction of global structure. This eliminates stereochemical losses and allows for flexible handling of complex bonding patterns in molecules.
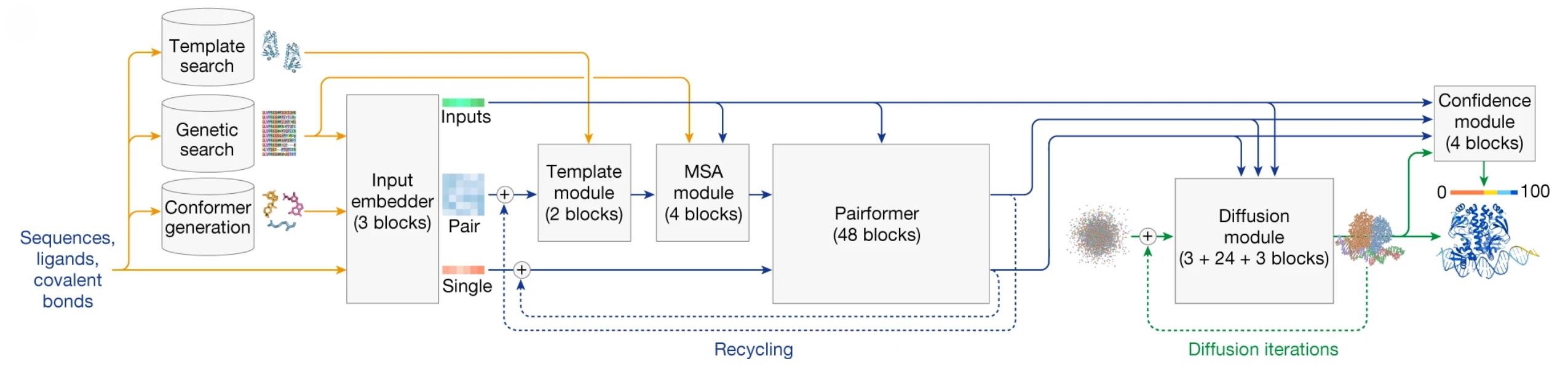 Figure 1. AF3 architecture: rectangles denote processing modules, arrows indicate data flow. Yellow for input data, blue for network activations, green for output data. Colored spheres represent physical atom coordinates. (Abramson J, et al., 2024)
Figure 1. AF3 architecture: rectangles denote processing modules, arrows indicate data flow. Yellow for input data, blue for network activations, green for output data. Colored spheres represent physical atom coordinates. (Abramson J, et al., 2024)
AF3's overall architecture retains core similarities to AF2 but introduces critical modifications that allow it to handle a wider variety of biomolecules while simplifying the architecture without sacrificing performance. Below is a detailed explanation of the AF3 process:
1. Tokenization Scheme
AF3 introduces an optimized tokenization scheme, where each amino acid residue corresponds to one token, and each nucleotide is treated similarly. For other molecular entities, each heavy atom is treated as a token. This system is more versatile, allowing AF3 to handle a variety of molecular types without excessive complexity.
2. Input Embedder
The Input Feature Embedder in AF3 encodes all the molecular information into a structured format. It processes chemical structures, assigns attention to all atoms, and outputs a single representation that captures the core features of all tokens (e.g., amino acids, nucleotides, and heavy atoms). This step sets the stage for accurate downstream modeling of molecular interactions.
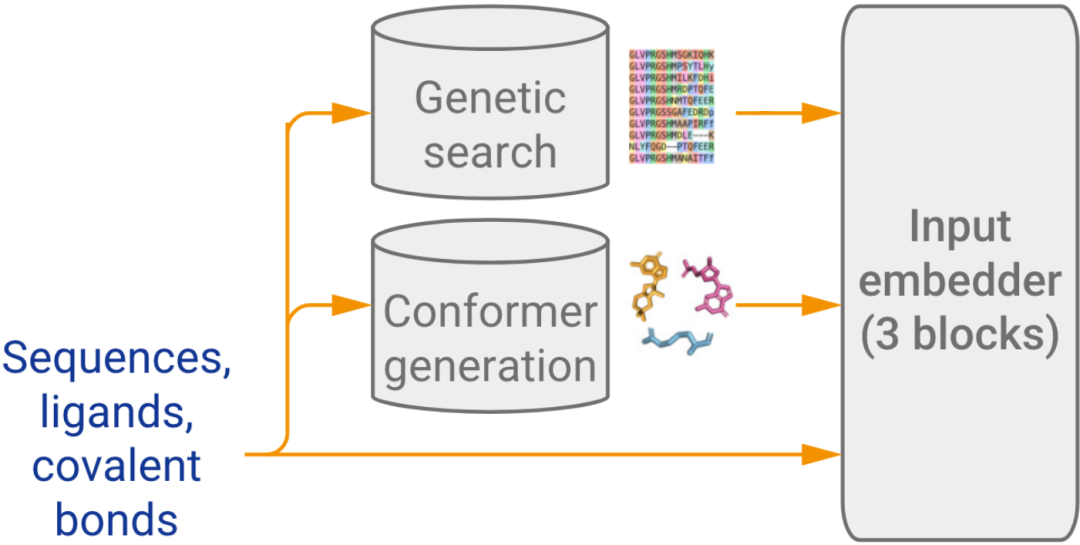 Figure 2. The part of the schematic representing the input embedder. (Abramson J, et al., 2024)
Figure 2. The part of the schematic representing the input embedder. (Abramson J, et al., 2024)
3. Pair Representation
The pair representation is constructed from the input features, similar to AF2. This representation captures the relationships between pairs of entities (proteins, nucleic acids, or other molecules) and is passed to a conditioning network, which recycles the data multiple times to refine the model.
4. Conditioning Network
The conditioning network in AF3 is composed of two primary modules: the Template Module and the MSA Module. The Template Module helps incorporate known structures (if available), while the MSA Module embeds multiple sequence alignment data into the pair representation. This process helps AF3 predict complex molecular structures by leveraging evolutionary information and known templates.
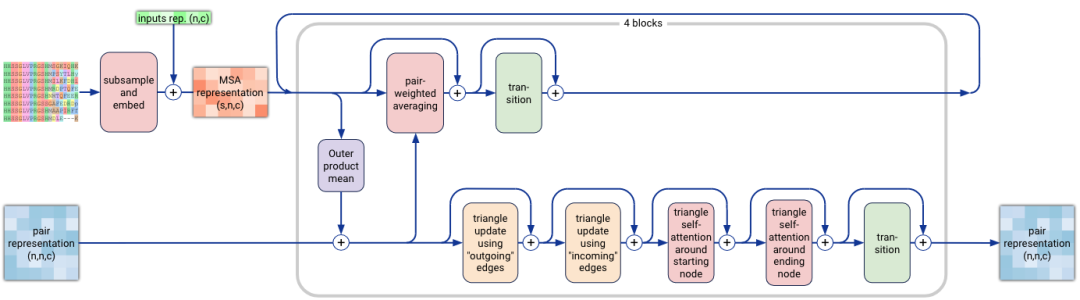 Figure 3. The multiple sequence alignment (MSA) module in AlphaFold3. (Abramson J, et al., 2024)
Figure 3. The multiple sequence alignment (MSA) module in AlphaFold3. (Abramson J, et al., 2024)
5. Pairformer Module
The Pairformer module is the centerpiece of AF3, replacing the Evoformer from AF2. It operates on the pair and single representations generated by the conditioning network. While pair representations capture molecular relationships, the single representation focuses on each individual molecular component. The Pairformer updates these representations and passes them to the next stage of the model.
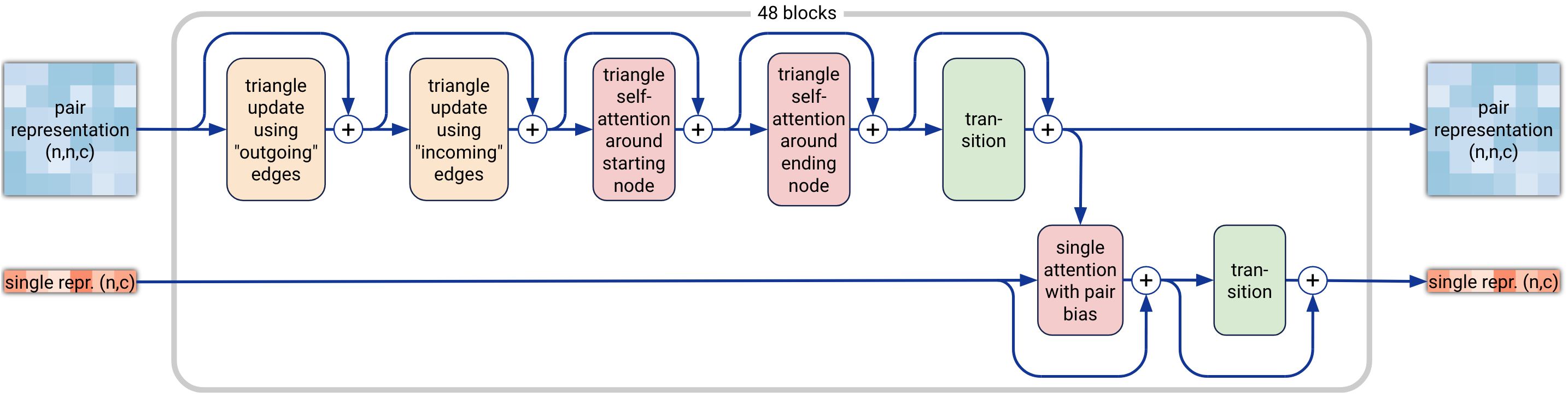 Figure 4. Pairformer module diagram, with inputs and outputs as paired and single representations, n for the number of tokens, and c for the channel count (paired at 128, single at 384). Each of the 48 blocks has a unique set of trainable parameters. (Abramson J, et al., 2024)
Figure 4. Pairformer module diagram, with inputs and outputs as paired and single representations, n for the number of tokens, and c for the channel count (paired at 128, single at 384). Each of the 48 blocks has a unique set of trainable parameters. (Abramson J, et al., 2024)
6. Diffusion Module
The Diffusion Module directly operates on raw atomic coordinates, using a diffusion process to predict the final structure. The multiscale nature of the diffusion process allows AF3 to learn protein structures at both local and global scales. At low noise levels, the model improves local stereochemistry, while higher noise enables the network to capture the global conformation of the biomolecule. This approach simplifies the architecture by avoiding the need for torsion-based parameterization of residues, as was required in AF2.
The diffusion model is conditioned using single and pair embeddings obtained from the Pairformer module, allowing the system to efficiently generate accurate molecular structures. AF3's use of diffusion modeling simplifies the prediction of general ligands and complex molecules.
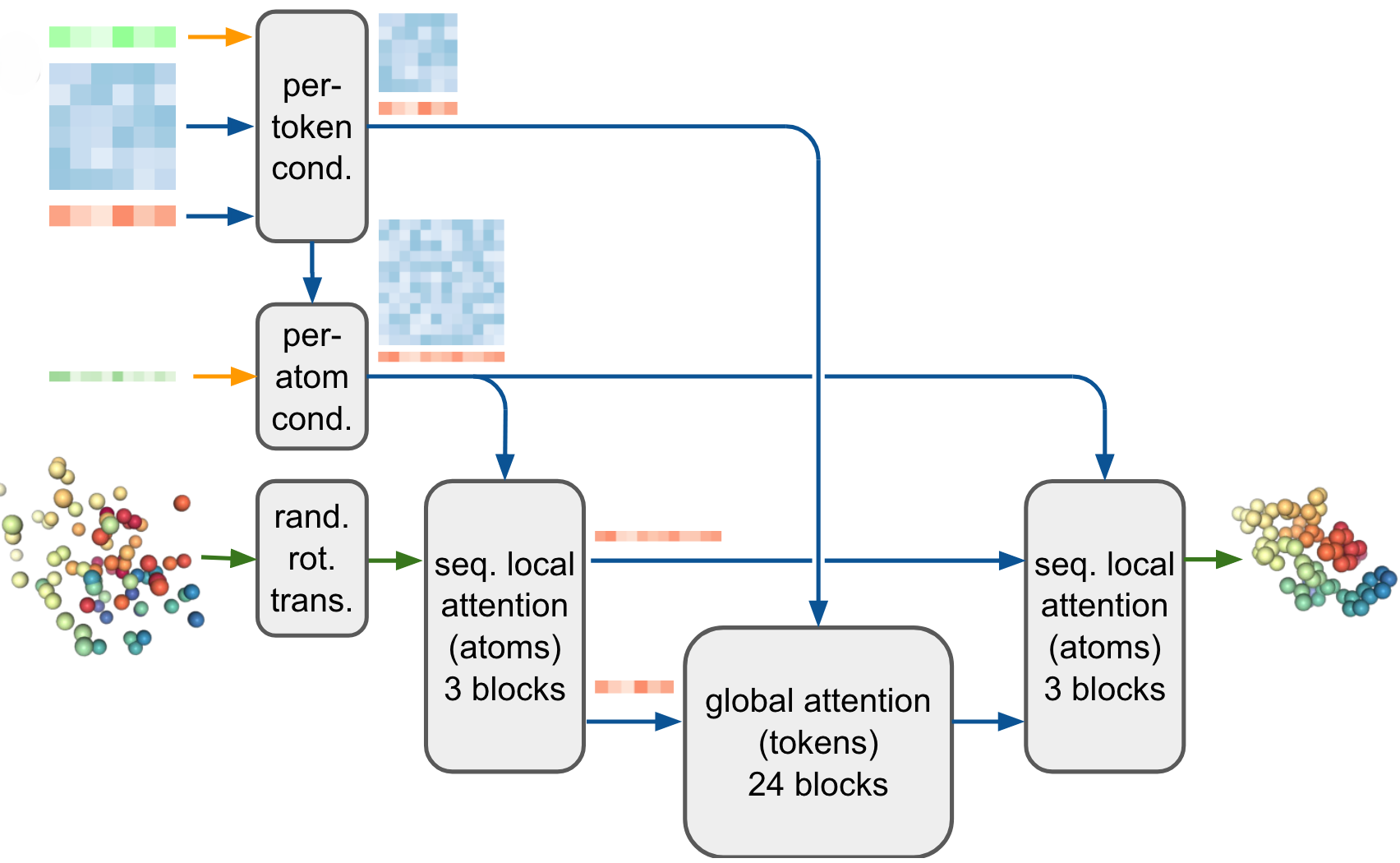 Figure 5. The diffusion module. Inputs: Per-token representations (green for input tokens; blue for paired tokens, orange for single tokens); per-atom conditions. Outputs: Colored spheres represent physical atomic coordinates. (Abramson J, et al., 2024)
Figure 5. The diffusion module. Inputs: Per-token representations (green for input tokens; blue for paired tokens, orange for single tokens); per-atom conditions. Outputs: Colored spheres represent physical atomic coordinates. (Abramson J, et al., 2024)
7. Confidence Module
AF3 concludes its prediction process with a Confidence Module, which calculates confidence scores for the predicted structures based on multiple inputs: pair representations, single representations, and structural outputs. The confidence head generates metrics such as the Predicted Local Distance Difference Test (pLDDT) and the Predicted Aligned Error (PAE) matrix, ensuring accurate confidence estimates across different prediction scales.
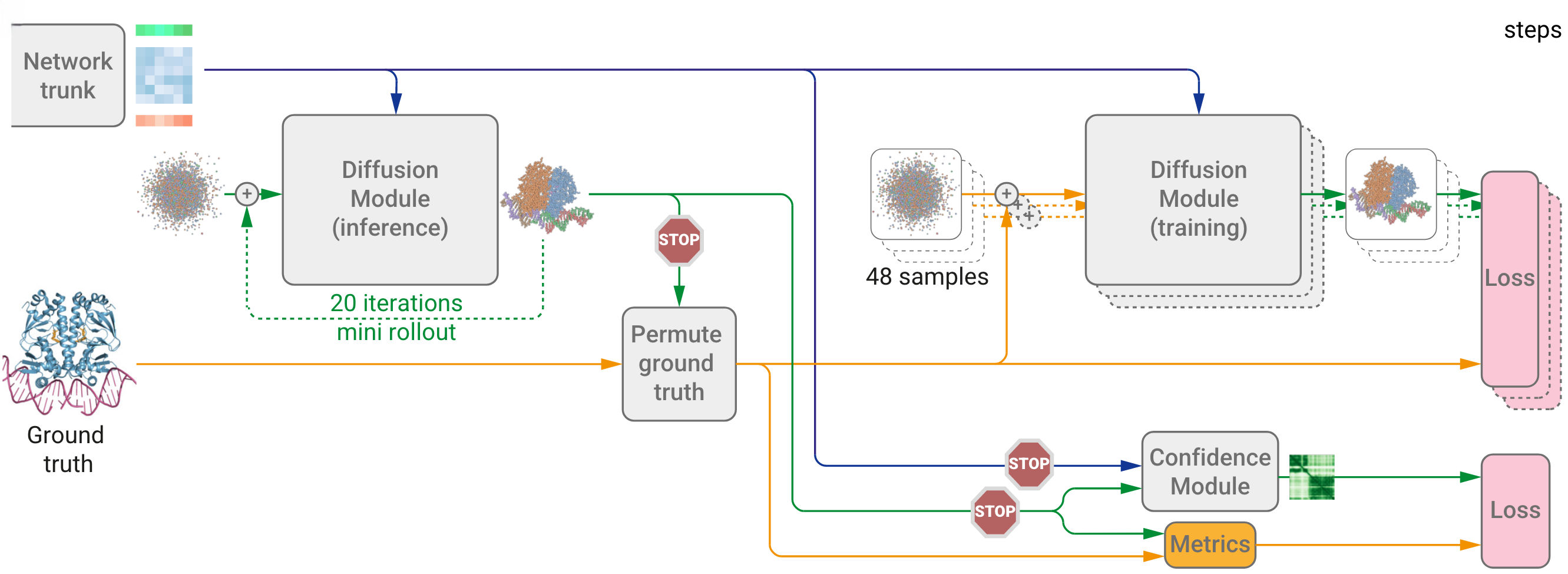 Figure 6. Training starts from the end of the network trunk. Green indicates input representations; blue for paired, orange for single. Blue arrows denote abstract activation arrays; yellow for real data; green for predicted data. The stop sign halts gradient flow. Both training and inference share weights across two diffusion modules. (Abramson J, et al., 2024)
Figure 6. Training starts from the end of the network trunk. Green indicates input representations; blue for paired, orange for single. Blue arrows denote abstract activation arrays; yellow for real data; green for predicted data. The stop sign halts gradient flow. Both training and inference share weights across two diffusion modules. (Abramson J, et al., 2024)
AlphaFold3 Model Predictive Performance
AF3 exhibits superior performance in predicting the structure of various biomolecular interactions, including protein-ligand complexes, protein-nucleic acid complexes, covalent modifications, and protein-protein interactions. This section will cover AF3's predictive accuracy in these key categories, showcasing its advantages over previous models.
1. Protein-Ligand Interactions
AF3 excels in predicting protein-ligand structures, outperforming specialized models in various molecular interaction types. Its performance was evaluated on the PoseBusters dataset, which contains 428 protein-ligand structures, including 161 structures added to the Protein Data Bank (PDB) after 2021. AF3 was trained on a separate dataset to avoid data leakage.
In terms of root mean square deviation (RMSD), AF3 demonstrated a significantly higher success rate (RMSD < 2Å) compared to Vina and RoseTTAFold All-Atom (RFAA) models. AF3's ability to predict accurate protein-ligand structures was evident in examples where traditional docking tools like Vina and Gold failed to dock successfully. AF3's performance on the PoseBusters v1 and v2 datasets also surpassed models like DiffDock and Uni-mol v2, making it the leading tool for ligand docking predictions.
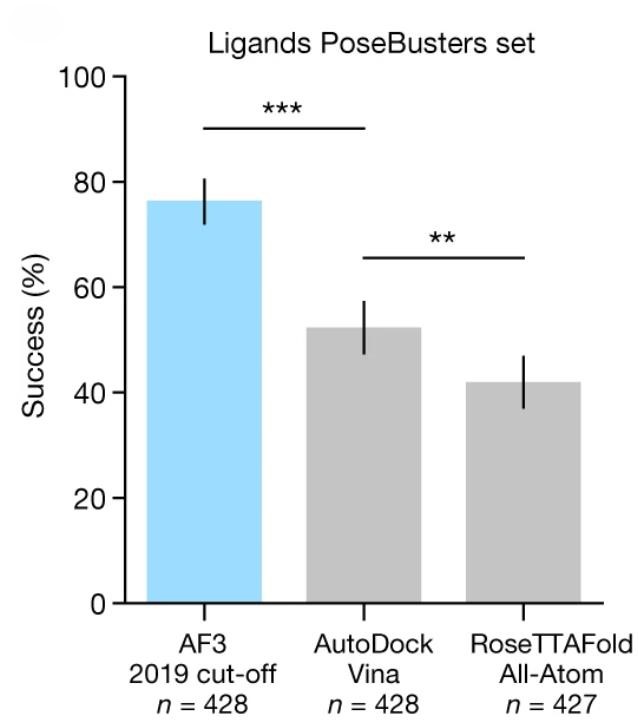 Figure 7. AF3 success rate on the PoseBusters dataset. Y-axis shows the percentage of ligands with RMSD < 2 Å, N denotes the number of targets. (Abramson J, et al., 2024)
Figure 7. AF3 success rate on the PoseBusters dataset. Y-axis shows the percentage of ligands with RMSD < 2 Å, N denotes the number of targets. (Abramson J, et al., 2024)
 Figure 8. AF3 can predict molecules that fail in Vina/Gold docking, with gray representing co-crystal structures, blue for predicted protein structures, and orange for predicted small molecule structures. (Abramson J, et al., 2024)
Figure 8. AF3 can predict molecules that fail in Vina/Gold docking, with gray representing co-crystal structures, blue for predicted protein structures, and orange for predicted small molecule structures. (Abramson J, et al., 2024)
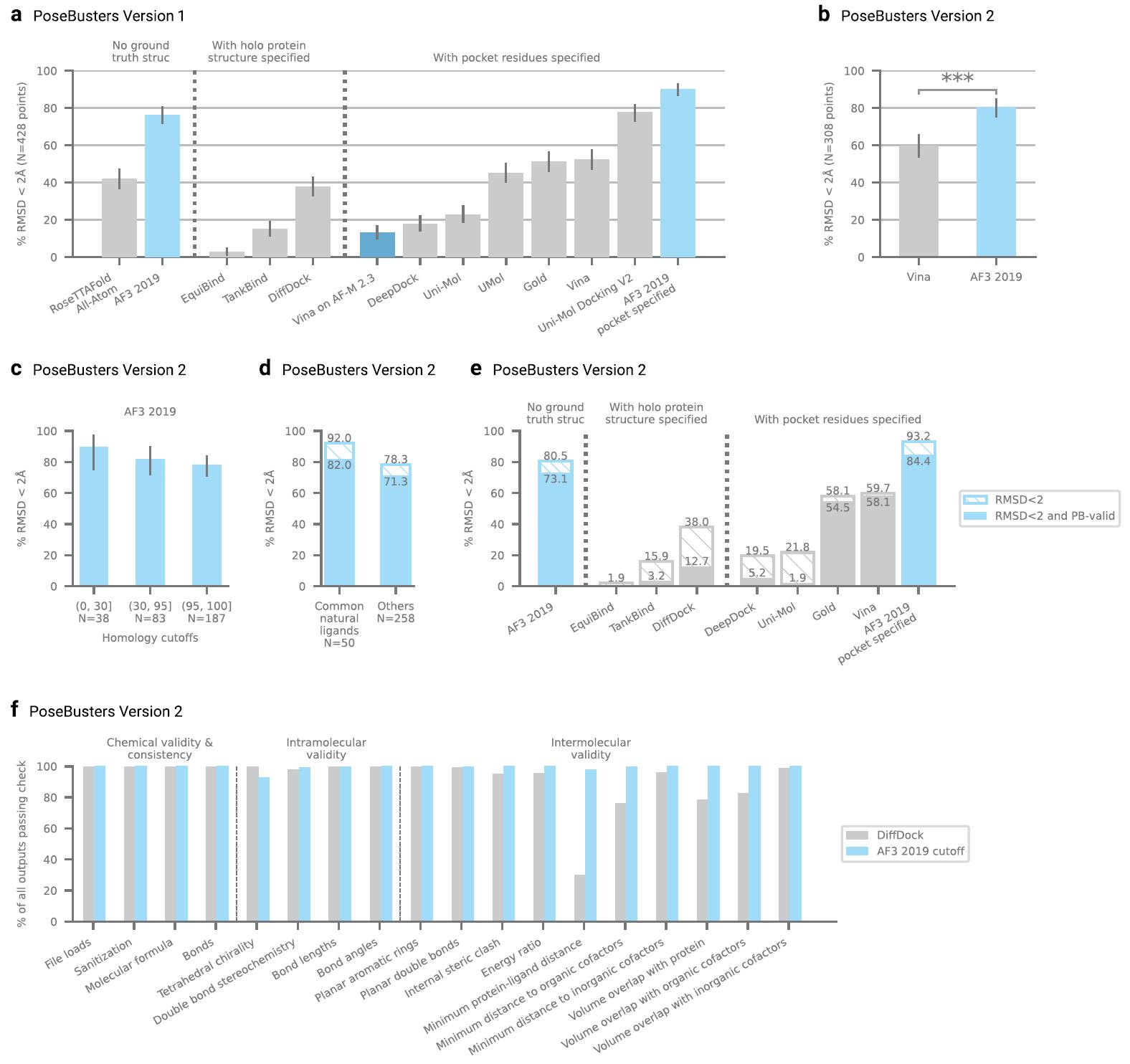 Figure 9. PoseBusters analysis. (Abramson J, et al., 2024)
Figure 9. PoseBusters analysis. (Abramson J, et al., 2024)
2. Protein-Nucleic Acid Complexes and RNA Monomers
AF3 outperforms RoseTTAFold2NA in predicting protein-nucleic acid complexes and RNA structures. While RFAA has lower accuracy compared to RoseTTAFold2NA, AF3 achieves a much higher success rate in protein-nucleic acid docking. AF3's ability to model large nucleic acid structures with thousands of residues gives it an edge over RoseTTAFold2NA, which is limited to smaller complexes.
Additionally, AF3 was evaluated on 10 RNA monomers from the CASP15 dataset. While AIchemy_RNA2 performed best overall in CASP15, AF3 still outperformed RoseTTAFold2NA in RNA structure prediction. This shows AF3's robust capability to handle diverse nucleic acid structures, making it a highly effective tool for RNA-based biological studies.
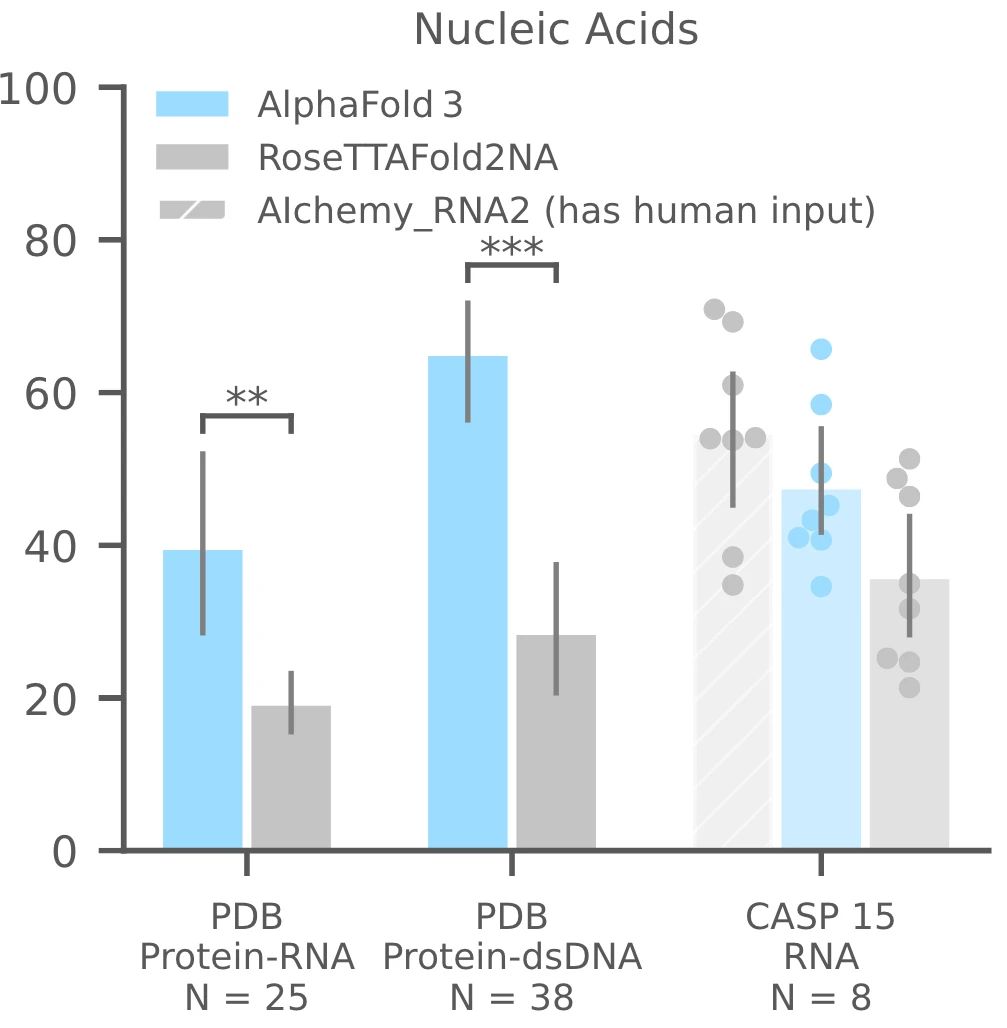 Figure 10. Protein-RNA and protein-double-stranded DNA in the PDB database; success rate of RNA monomers in the CSAP15 competition. Success rate is based on interface LDDT for complexes, and LDDT for individual RNA monomers. N represents the number of targets. (Abramson J, et al., 2024)
Figure 10. Protein-RNA and protein-double-stranded DNA in the PDB database; success rate of RNA monomers in the CSAP15 competition. Success rate is based on interface LDDT for complexes, and LDDT for individual RNA monomers. N represents the number of targets. (Abramson J, et al., 2024)
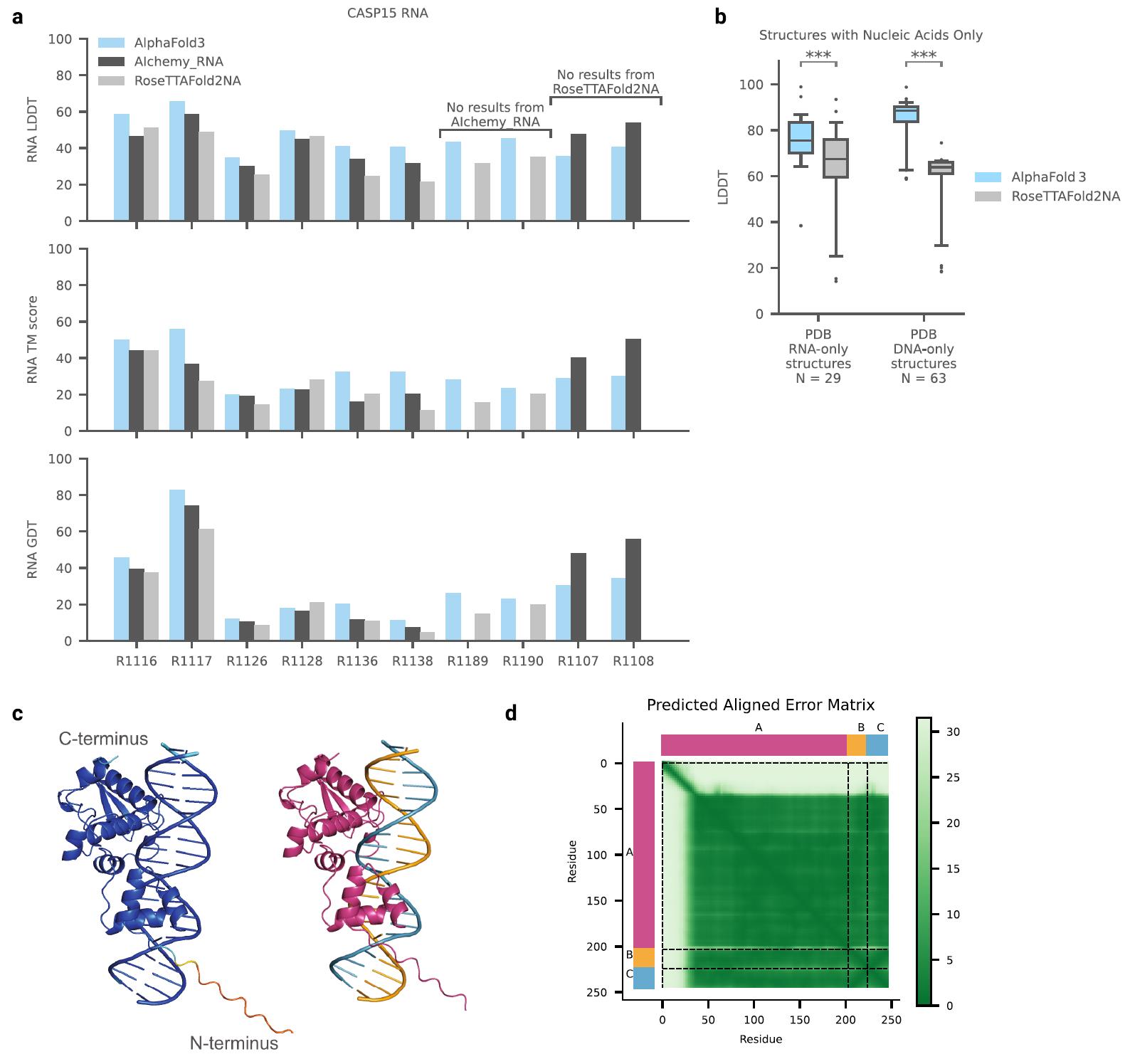 Figure 11. Nucleic acid prediction accuracy and confidences. (Abramson J, et al., 2024)
Figure 11. Nucleic acid prediction accuracy and confidences. (Abramson J, et al., 2024)
3. Covalent Modifications
AF3 can also accurately predict covalent modifications, including bonded ligands, glycosylation, and modifications to protein and nucleic acid residue. These modifications include changes to any polymer residue, whether protein, RNA, or DNA. AF3 defines success in this domain as RMSD < 2Å, and its accuracy in predicting phosphorylation and other modifications makes it superior to models like AF2. These predictions help in understanding the biochemical effects of modifications, such as phosphorylation, on molecular structure and function.
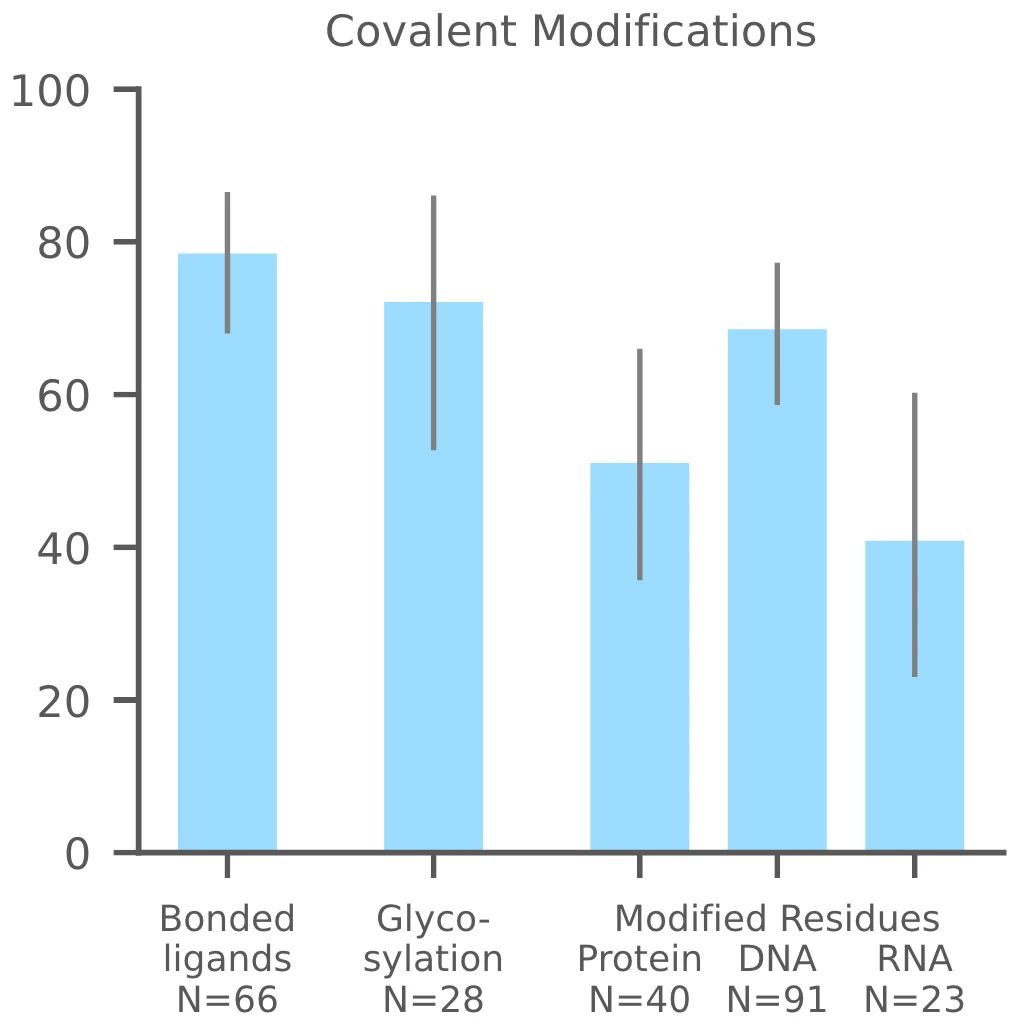 Figure 12. Y-axis: Success rate defined as the percentage of ligands with RMSD < 2 Å, N denotes the number of targets. Categories include: protein-ligand and small molecules, protein glycosylation, protein modification, DNA/RNA modification. (Abramson J, et al., 2024)
Figure 12. Y-axis: Success rate defined as the percentage of ligands with RMSD < 2 Å, N denotes the number of targets. Categories include: protein-ligand and small molecules, protein glycosylation, protein modification, DNA/RNA modification. (Abramson J, et al., 2024)
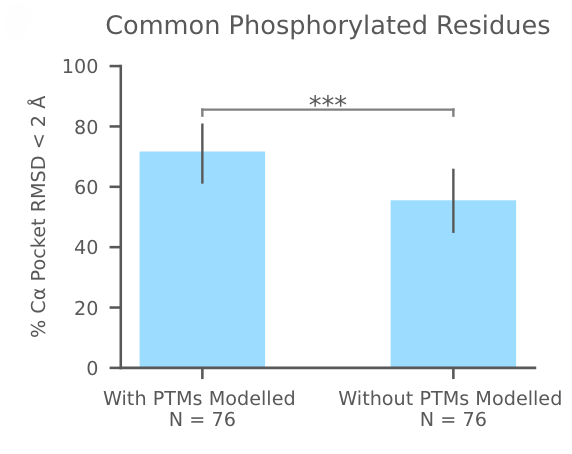 Figure 13. In phosphorylation scenarios (SEP, TPO, PTR, NEP, HIP), the prediction success rate of AF3, where PTM stands for post-translational modification. (Abramson J, et al., 2024)
Figure 13. In phosphorylation scenarios (SEP, TPO, PTR, NEP, HIP), the prediction success rate of AF3, where PTM stands for post-translational modification. (Abramson J, et al., 2024)
4. Protein-Protein Interactions and Protein Monomer
AF3 has significantly improved the prediction of protein complexes and monomers compared to AlphaFold-Multimer v2.3. Its performance in antibody-protein interactions has been particularly impressive, showcasing a marked improvement in accuracy. AF3's predictions of protein monomers also show a clear enhancement in LDDT scores, a measure of prediction accuracy.
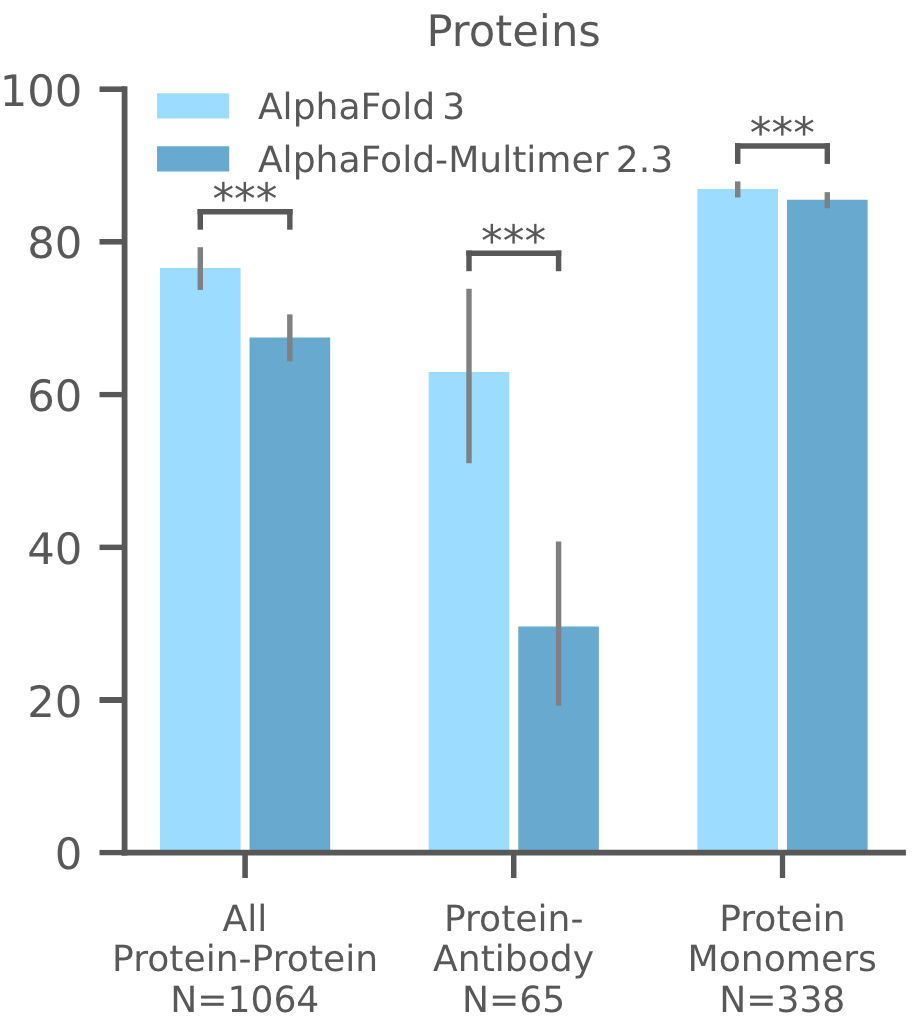 Figure 14. Prediction success rates for protein multimers, protein-antibody complexes, and protein monomers in the Recent PDB evaluation set. Success for protein multimers and protein-antibody complexes is defined as DockQ > 0.23. Success for protein monomers is defined by the LDDT metric. N represents the number of targets. (Abramson J, et al., 2024)
Figure 14. Prediction success rates for protein multimers, protein-antibody complexes, and protein monomers in the Recent PDB evaluation set. Success for protein multimers and protein-antibody complexes is defined as DockQ > 0.23. Success for protein monomers is defined by the LDDT metric. N represents the number of targets. (Abramson J, et al., 2024)
Correlation Between Accuracy and Confidence Metrics
One of the hallmark features of AlphaFold3 is the strong correlation between its accuracy and confidence metrics. The confidence scores are calibrated based on the predicted LDDT and Predicted Aligned Error (PAE) values, ensuring that higher confidence scores directly translate to more reliable predictions. This aspect allows researchers to effectively filter out lower-quality predictions, improving the overall accuracy and utility of the model.
AF3's ipTM scores (Interface-predicted TM scores) also correlate well with DockQ scores for protein-protein and protein-nucleic acid interfaces. The system's confidence tracking mechanism has become a critical tool in assessing the reliability of complex predictions, particularly for ligand-binding sites and antibody-antigen interfaces.
AlphaFold3 Model Limitations
Despite the significant advancements made by AF3 in protein structure prediction, several limitations persist, demonstrating the challenges in computational modeling and the reality that no single approach can excel in all scenarios. Below is a comprehensive overview of AF3's key limitations:
1. Stereochemistry Limitations
One of the most notable challenges faced by AF3 is related to stereochemistry:
- Chirality Issues: Even when provided with correct reference structures for chirality, AF3 occasionally outputs models that violate chirality. In the PoseBusters benchmark, AF3 incorporated penalties for chirality violations, yet the chirality violation rate remained at 4.4%.
- Atomic Overlap: In certain cases, especially in protein-nucleic acid complexes, AF3 may produce atomic overlaps where atoms are unrealistically placed in the same space. Although penalties were applied to mitigate these overlaps, this issue has not been entirely eliminated.
2. Hallucination Problem
The transition from the non-generative AF2 model to the diffusion-based AF3 introduced the issue of hallucinated structures in disordered regions of proteins. While AF3 does predict these regions with low confidence, the disordered areas lack the characteristic ribbon-like appearance generated by AF2. To address this, AF3 adopted distillation training using AF2 predictions and introduced penalties that encourage greater solvent-accessible surface area to reduce hallucination. However, this challenge remains a limitation when predicting inherently disordered protein regions.
3. Dynamics of Biomolecular Systems
A fundamental limitation of AF3 is its focus on predicting static structures, which do not capture the dynamic behavior of biomolecules in solution. Biomolecules are highly dynamic and undergo conformational changes in response to their environment or interactions with ligands. AF3 is limited to predicting a single static state, even though it uses diffusion and multiple random seeds. Thus, the model struggles to approximate the ensemble of possible conformations seen in reality, such as those exhibited during protein-ligand interactions or signal transduction pathways.
4. Accuracy Issues with Specific Targets
For certain targets, AF3 may fail to predict the correct conformational states. For example, in the case of E3 ubiquitin ligases, AF3 tends to predict only the closed state (bound to ligands) and fails to model the open apo state (when the protein is unbound). This indicates that AF3 may struggle with accurately modeling systems that naturally switch between multiple conformational states, limiting its utility in scenarios where conformational flexibility is crucial.
5. Accuracy and Computational Costs
While AF3 can produce highly accurate predictions, achieving these results often requires generating and evaluating a large number of model predictions. For instance, in antibody-antigen complex predictions, increasing the number of random seeds improves prediction quality, but it also demands more computational resources. This trade-off between accuracy and computational cost presents a challenge, especially for large-scale predictions where high-throughput modeling is needed.
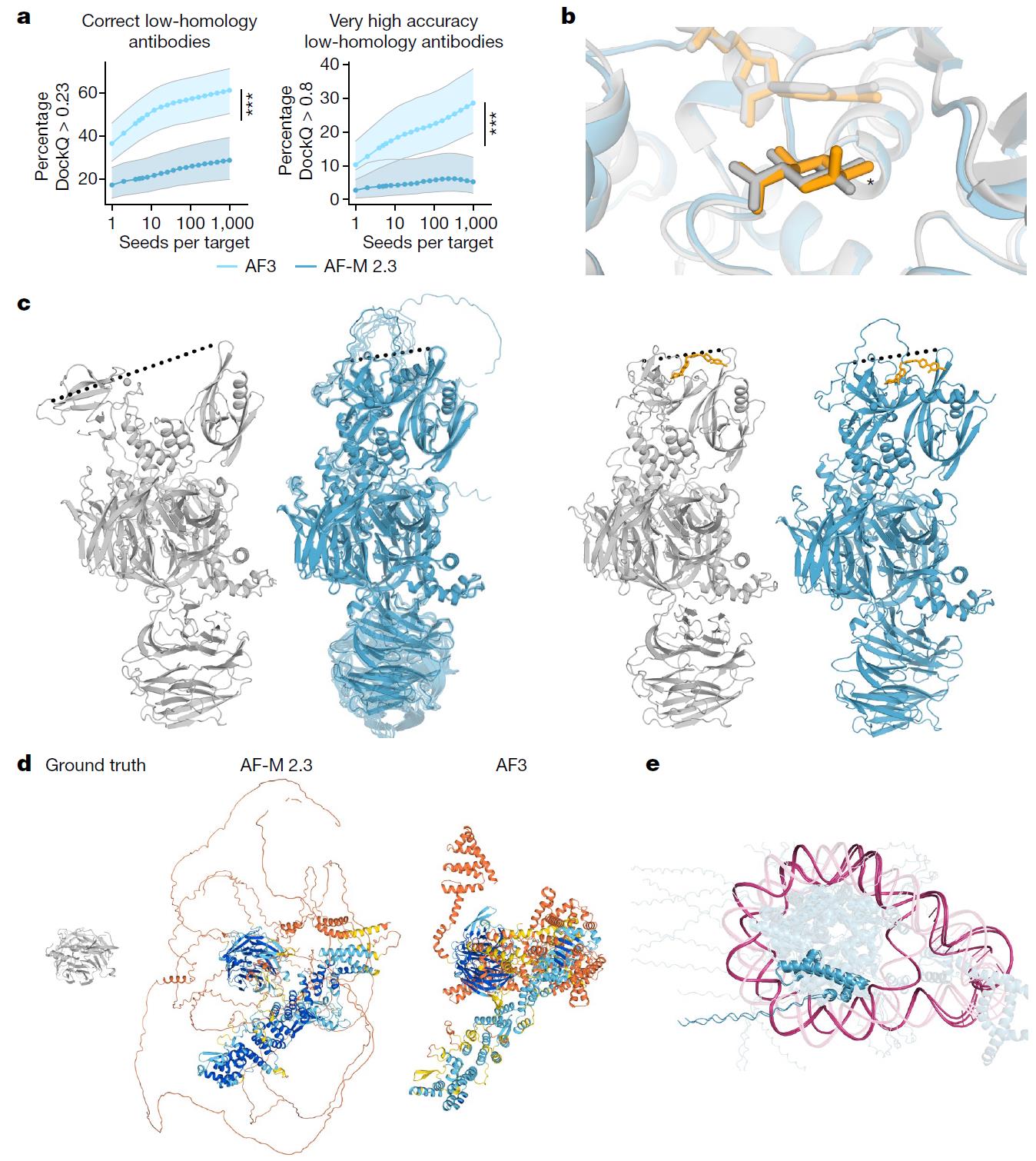 Figure 15. AlphaFold3 Limitations. a: The quality of antibody predictions increases with the number of model seeds. b: Stereochemistry prediction errors. c: Limitations in conformational coverage. d: Unresolved residues in nuclear pore complexes. e: Erroneous predictions of the tri-nucleosome. (Abramson J, et al., 2024)
Figure 15. AlphaFold3 Limitations. a: The quality of antibody predictions increases with the number of model seeds. b: Stereochemistry prediction errors. c: Limitations in conformational coverage. d: Unresolved residues in nuclear pore complexes. e: Erroneous predictions of the tri-nucleosome. (Abramson J, et al., 2024)
Creative Biostructure is committed to providing comprehensive structural analysis services to make up for the shortcomings of AlphaFold3 in structural prediction. We combine experimental data and prediction results to provide more accurate structural models and determine the true structure of proteins.
We provide dynamic conformation analysis services, which capture multiple conformations of proteins in different environments through experimental methods, including conformational changes when interacting with ligands.
In addition, we also provide guidance services for protein engineering and drug discovery, using AlphaFold3's prediction results as a starting point to accelerate the drug design and development process through experimental verification and optimization. Our services are designed to provide customers with a one-stop solution from prediction to experimental verification. For further details about our services and solutions, don't hesitate to contact us.
References
- Senior A W, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020. 577(7792): 706-710.
- Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021. 596(7873): 583-589.
- Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024: 1-3.