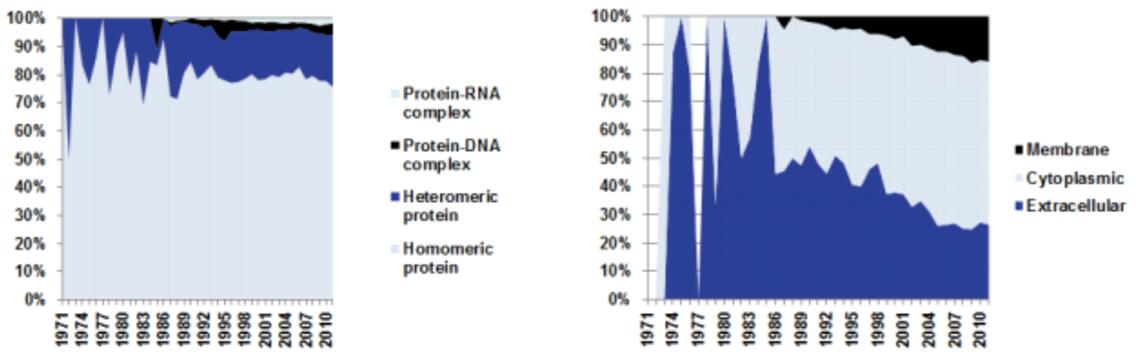
The evolution from homology modeling to advanced deep learning models like AlphaFold represents a transformative paradigm shift in structural biology. This shift has enabled accurate, large-scale protein structure prediction, revolutionizing how researchers approach protein-related diseases, drug discovery, and the study of fundamental biological processes.
Importance of Protein Structure Prediction
In the field of biology, proteins are essential molecular machines that drive virtually every biochemical process. The function of a protein is intrinsically tied to its three-dimensional structure, which determines how it interacts with other molecules and performs its role in the cell. Accurate protein structure prediction is critical not only for understanding the fundamental mechanisms of life but also for practical applications in medicine and drug development. By knowing a protein's structure, scientists can design drugs that fit perfectly into the protein's active sites or disrupt harmful protein-protein interactions, thereby offering therapeutic solutions for diseases.
Historical Context of Protein Structure Determination
The determination of protein structures has historically relied on experimental techniques such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy and cryo-electron microscopy (Cryo-EM). In 1957, John C. Kendrew and Max F. Perutz resolved the first protein structures using X-ray crystallography, a groundbreaking achievement in structural biology. Soon after, Christian B. Anfinsen Jr. proposed that the three-dimensional structure of a protein is determined by its amino acid sequence, a hypothesis that set the foundation for future studies in protein folding. However, experimental methods have always been labor-intensive, expensive, and limited in scope, making the search for computational methods essential for large-scale protein structure prediction.
Homology Modeling
What is Homology Modeling?
Homology modeling, also known as comparative modeling, is one of the earliest and most widely used computational techniques for predicting protein structures. The principle behind homology modeling is based on the observation that proteins with similar sequences tend to adopt similar three-dimensional structures. By aligning the target protein sequence to a known template structure, researchers can predict the structure of the unknown protein. This method was particularly useful in the early stages of computational biology when template structures were sparse.
Techniques and Algorithms Used in Homology Modeling
The process of homology modeling involves several key steps:
- Template Identification: A sequence alignment algorithm identifies a template protein structure with significant similarity to the target sequence.
- Alignment: The target sequence is aligned to the template structure using advanced algorithms like BLAST or PSI-BLAST.
- Model Building: A 3D model of the target protein is constructed by copying the backbone and side chain positions from the template.
- Refinement: The model undergoes energy minimization to optimize bond angles, torsions, and minimize clashes.
- Validation: Tools like PROCHECK or Ramachandran plots are used to evaluate the model's quality.
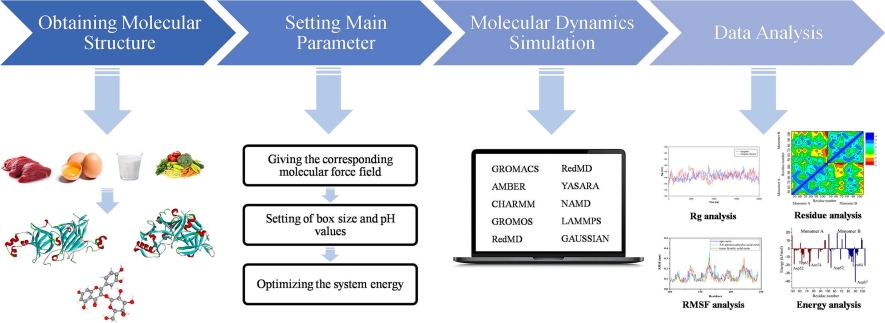
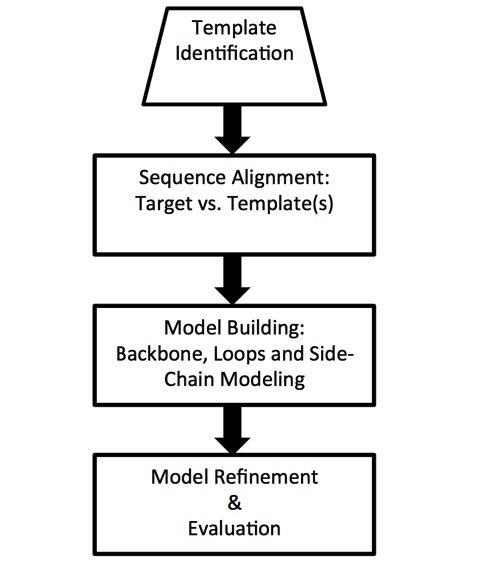 Figure 1. The
main steps in homology modeling. (Hasani H J, et al., 2017)
Figure 1. The
main steps in homology modeling. (Hasani H J, et al., 2017)
Successes and Limitations of Homology Modeling
Homology modeling has been successful in predicting the structures of proteins that share high sequence identity with known templates. It has been particularly useful in early drug discovery efforts, where researchers could model protein-drug interactions for therapeutically relevant proteins. However, homology modeling has inherent limitations. It relies heavily on the availability of homologous structures in protein databases, and as sequence identity decreases, the accuracy of the model declines. Additionally, homology modeling cannot accurately predict novel protein folds, limiting its utility for proteins with no known structural relatives.
Computational Methods: The Rise of Ab Initio and Threading
Ab Initio Methods
Ab initio methods represent a fundamentally different approach from homology modeling, as they predict protein structures from physical principles without relying on homologous templates. These methods attempt to predict the folding of proteins solely based on their amino acid sequences by simulating the physical forces that drive folding, such as hydrophobic interactions, van der Waals forces, and hydrogen bonding. However, due to the vast number of possible conformations a protein can adopt, ab initio methods have been computationally demanding and historically limited in accuracy.
Threading Methods
Threading methods, or fold recognition, attempt to fit a target sequence directly onto a set of known protein folds, regardless of sequence similarity. Threading leverages structural templates and identifies the most compatible structure based on physical and statistical factors. This method is useful when homology modeling fails due to a lack of sequence similarity but there are structurally similar folds available.
Comparison of Ab Initio and Threading with Homology Modeling
While homology modeling relies on sequence similarity, ab initio and threading methods explore broader structural predictions. Ab initio approaches offer the potential to predict entirely novel folds, whereas threading can identify distant structural relatives. Both methods are computationally more intensive and historically less accurate than homology modeling, but they offer solutions when homology-based approaches are not feasible.
Both ab initio and threading methods faced challenges due to computational complexity and the lack of high-resolution results. However, the introduction of deep learning and AI-driven approaches marked a significant breakthrough, as these models could learn intricate protein folding patterns from large datasets, setting the stage for AlphaFold's development.
Development of AlphaFold
Underlying Technology of AlphaFold
Deep learning has transformed bioinformatics by enabling the analysis of vast amounts of biological data with unprecedented accuracy. Deep learning models, particularly convolutional neural networks (CNNs) and transformers, can capture complex patterns in data and apply these insights to tasks like protein structure prediction. By training on large datasets of protein sequences and structures, these models have dramatically improved the accuracy of predictions.
AlphaFold utilized neural networks to predict protein structures with atomic-level accuracy. Early versions, like AlphaFold1, employed relatively simple architectures, but the introduction of AlphaFold2 brought the transformative power of the transformer architecture. This allowed the system to integrate evolutionary, physical, and biological information, producing highly accurate structural predictions.
AlphaFold2 integrates a novel architecture combining multiple sequence alignments (MSAs) with a transformer-based system that processes pairwise residue interactions. The Evoformer module refines this information, and the structure module predicts atomic coordinates. In AlphaFold3, the diffusion module replaced the structure module, further enhancing its ability to predict complex biological assemblies, including protein-ligand and protein-DNA interactions.
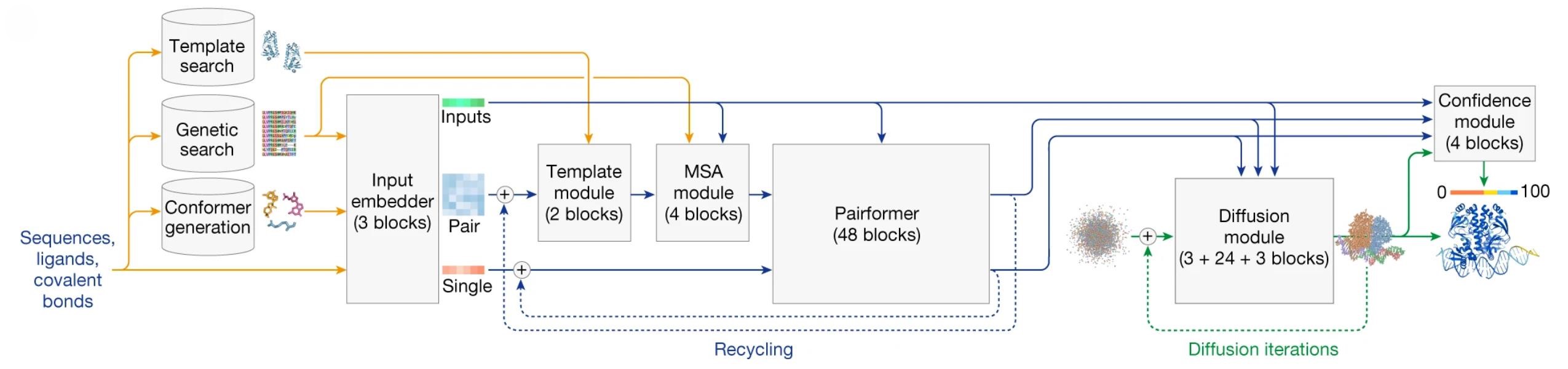 Figure 2. AlphaFold3 inference architecture. Rectangles denote processing modules; arrows indicate data flow. Colors: Yellow - input data, Blue - abstract network activations, Green - output data. Colored spheres represent
physical atom coordinates. (Abramson J, et al., 2024)
Figure 2. AlphaFold3 inference architecture. Rectangles denote processing modules; arrows indicate data flow. Colors: Yellow - input data, Blue - abstract network activations, Green - output data. Colored spheres represent
physical atom coordinates. (Abramson J, et al., 2024)
AlphaFold Database
The AlphaFold database is a comprehensive resource that provides open access to the predicted structures of hundreds of thousands of proteins. By offering high-quality structural predictions, it has democratized access to protein structure information, allowing researchers worldwide to explore the 3D shapes of proteins without the need for expensive experimental techniques.
The availability of the AlphaFold database has major implications for both basic research and applied sciences. It accelerates the identification of drug targets, and enhances our understanding of disease-related proteins.
Applications of AlphaFold in Research and Medicine
- Drug Discovery and Development: AlphaFold has become a game-changer in drug discovery. Its ability to predict protein structures helps identify potential drug-binding sites, design more effective inhibitors, and reduce the time required for lead optimization.
- Understanding Protein-Protein Interactions: Protein-protein interactions are fundamental to many biological processes and diseases. AlphaFold's accuracy in predicting these interactions provides crucial insights into molecular mechanisms that were previously difficult to study. These predictions facilitate the design of drugs that can modulate such interactions, which is critical for treating diseases like cancer and autoimmune disorders.
- Insights into Disease Mechanisms and Potential Therapeutic Targets: AlphaFold enables researchers to study the structural basis of disease-related proteins, shedding light on previously unknown mechanisms. For example, AlphaFold3 has been used to predict interactions between viral spike proteins and human antibodies, providing crucial insights for vaccine design.
Challenges of AlphaFold and Deep Learning in Protein Structure Prediction
Despite its successes, AlphaFold still has limitations, such as occasional stereochemistry errors and challenges in modeling protein dynamics. Additionally, its ability to predict multi-subunit assemblies and protein-RNA interactions is still being refined.
- Experimental Validation: Combining AlphaFold with methods like cryo-electron microscopy and NMR ensures more robust structural models.
- Future Directions: Focus on improving predictions for protein complexes, dynamic structures, and interactions with small molecules. AlphaFold's framework may inspire models for other biological systems like carbohydrates and lipids.
- Interdisciplinary Collaboration: Collaboration between biologists, computational scientists, chemists, and physicists is essential for progress in protein structure prediction.
At Creative Biostructure, we emphasize the importance of integrating computational and experimental methods for comprehensive structural analysis. By establishing interdisciplinary partnerships, we support customers to integrate AlphaFold predictions into workflows to accelerate structural biology research and drug discovery.
In addition, we can efficiently use the structural information output by AlphaFold3 as a guide for more precise protein modification, thereby greatly improving the efficiency and success rate of protein modification and meeting the diverse needs of various proteins. If you are interested in our services and products, please feel free to contact us for more information.
References
- Hasani H J, Barakat K. Homology modeling: an overview of fundamentals and tools. Int. Rev. Model. Simul. 2017. 10(2): 1-14.
- Senior A W, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020. 577(7792): 706-710.
- Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021. 596(7873): 583-589.
- Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024: 1-3.
- Callaway E. Major AlphaFold upgrade offers boost for drug discovery. Nature. 2024. 629(8012): 509-510.