Cryo-electron microscopy (cryo-EM) has revolutionized the world of structural biology by allowing researchers to visualize macromolecular complexes at atomic resolution. In particular, single particle analysis (SPA) has emerged as a powerful technique for determining structures of large, flexible biomolecules. However, to fully unlock the potential of cryo-EM, the data processing pipeline plays a critical role. This article will guide you through the entire single particle cryo-EM data processing workflow, highlight essential cryo-EM software solutions, and showcase case studies.
Introduction to Single Particle Cryo-EM Data Processing
Single Particle Analysis vs. Other Cryo-EM Techniques
Single particle analysis (SPA) is one of the most widely used cryo-EM techniques, where individual particles (usually proteins or complexes) are isolated, imaged, and computationally combined to generate a high-resolution 3D structure. This contrasts with other cryo-EM methods like cryo-ET (cryo-electron tomography), which is often used for studying cellular structures and macromolecular complexes in situ. SPA is particularly powerful when studying molecules in solution, as it captures a broader range of conformational states.
Importance of Data Processing in Single Particle Analysis
The success of SPA is heavily dependent on the quality of the data processing pipeline. Without accurate data processing techniques, even the most high-quality cryo-EM images can yield poor or unusable 3D reconstructions. Data processing steps such as motion correction, contrast transfer function (CTF) estimation, and particle classification are all critical in obtaining reliable and high-resolution structures.
Select Service
Related Reading
- Overview of Cryo-Electron Microscopy (Cryo-EM) Technology
- Single Particle Cryo-EM Overview
- Overview of Cryo-Electron Tomography (Cryo-ET) Technology
- MicroED Technology Overview
- Single Particle Cryo-EM vs Cryo-ET
- MicroED vs Cryo-EM SPA: Key Differences and Applications
- Cryo-ET Data Processing: Workflow, Advances, and Software Tools
Single Particle Cryo-EM Data Processing Workflow
1. Data Acquisition and Initial Processing
The first step in any cryo-EM project is data acquisition. Using advanced detectors like direct electron detectors (DEDs), researchers can capture high-quality images of particles. However, effective data acquisition is just the beginning. The initial processing of raw images is essential to prepare them for further analysis. Tools such as EPU are commonly used for efficient data collection and initial image processing.
2. Motion Correction and CTF Estimation
Motion correction is an essential step in cryo-EM data processing. During the imaging process, particles can move due to the beam-induced motion, leading to blurry images. Motion correction tools, like those found in RELION, can accurately correct these motions to restore image clarity.
CTF estimation is another crucial step. The CTF corrects for distortions introduced by the microscope's optics. Accurate CTF correction is necessary to ensure the high-quality reconstruction of the particle's 3D structure.
3. Particle Picking and Extraction
Particle picking is the process of identifying individual particles within the cryo-EM micrographs. There are two main strategies: manual picking, which requires human intervention, and automated particle picking, which leverages algorithms to identify particles. While automated techniques are faster, they often require manual validation to ensure accuracy.
4. 2D and 3D Classification
Once particles are picked and extracted, they undergo 2D classification to identify high-quality particles from noisy data. After 2D classification, the best particles are used for 3D classification, which helps to refine the structural model by grouping particles into different orientations.
5. Ab Initio 3D Reconstruction
In this step, an initial 3D model of the molecule is generated using the classified particles. This is typically done using ab initio methods that rely on the alignment of particles to build an initial 3D structure.
6. High-Resolution Refinement
After the initial 3D model is generated, the structure undergoes high-resolution refinement to improve its quality. During refinement, algorithms optimize the alignment and orientations of the particles to achieve the best possible resolution.
7. Heterogeneity Analysis
Heterogeneity analysis is a powerful technique used to account for variations in the particles. This step is particularly useful for studying flexible or multi-subunit proteins. By identifying distinct conformational states within the particle population, researchers can obtain a more accurate representation of the biological system.
Cryo-EM Software Solutions for Single Particle Analysis: Open-Source Tools
1. RELION
RELION (REgularized LIkelihood Optimized Reconstruction) is one of the most widely used open-source software packages for cryo-EM single particle analysis. It has been developed by the MRC Laboratory of Molecular Biology (LMB) and is continuously updated by an active open-source community.
| Key Features |
|
| Pros |
|
| Cons |
|
2. Cryo-EMSPIN
Cryo-EMSPIN is an open-source package designed to facilitate large-scale cryo-EM data processing and analysis. It is primarily used for efficient particle recognition, extraction, and reconstruction in cryo-EM single particle analysis.
| Key Features |
|
| Pros |
|
| Cons |
|
3. Scipion
Scipion is another open-source software designed to integrate multiple cryo-EM software tools into one unified platform. It serves as a wrapper for various cryo-EM programs, including RELION, SPIN, and others, making it an excellent choice for researchers who want to use multiple tools in their workflow.
| Key Features |
|
| Pros |
|
| Cons |
|
4. SPIN (Single Particle Imaging and Navigation)
SPIN is an open-source cryo-EM software suite focused on the automated processing of cryo-EM data, particularly for large-scale projects. SPIN is designed to handle the complexities of single particle analysis in high-throughput scenarios.
| Key Features |
|
| Pros |
|
| Cons |
|
5. Xmipp
Xmipp is an open-source software package focused on cryo-EM image processing and single particle analysis. Developed by the CIC bioGUNE in Spain, it provides a wide range of tools for cryo-EM data processing, from initial particle picking to final reconstruction.
| Key Features |
|
| Pros |
|
| Cons |
|
While there are many cryo-EM software solutions available, choosing the right one depends on factors such as the complexity of the sample, the size of the dataset, and the desired resolution. Comparing features, ease of use, and support services can help determine the best platform for each specific project.
Cryo-EM Image Processing Techniques in Single Particle Analysis
Cryo-EM image processing is crucial for achieving high-quality reconstructions. The process involves several advanced techniques to address challenges like low signal-to-noise ratios (SNR), motion artifacts, and particle heterogeneity. Below are some key techniques and solutions for enhancing image quality in single particle analysis.
Advanced Algorithms for Image Enhancement
- Fourier Interpolation: Enhances image resolution by improving particle alignment in the frequency domain. This technique helps reduce blurring and increases signal quality, especially in poorly defined regions.
- Particle Polishing: Refines particle images by aligning them in both real and reciprocal space. This improves particle position accuracy and compensates for distortions caused by sample movement during data collection.
These algorithms work together to improve image clarity and facilitate high-resolution reconstructions.
Machine Learning and AI in Image Processing
- Particle Detection: AI-based methods automate particle picking, improving accuracy and efficiency. These techniques help identify particles in noisy or complex datasets.
- Particle Classification: Machine learning optimizes particle sorting, ensuring higher-quality 2D and 3D classifications and leading to more accurate final reconstructions.
- Denoising: AI reduces background noise in cryo-EM images, enhancing signal clarity and resolution in the final structure.
Machine learning and AI play a vital role in speeding up the cryo-EM workflow while enhancing the precision of data analysis. Learn more in How AI is Transforming Cryo-EM Data Analysis.
Challenges and Solutions in Image Quality
| Challenges | Solutions |
|---|---|
| Low Signal-to-Noise Ratio (SNR) | Cryo-EM data often suffers from low SNR. Solutions include the use of direct electron detectors that offer greater sensitivity and faster image capture, improving data quality. |
| Motion Artifacts | Motion correction algorithms (like MotionCor2) compensate for sample drift, ensuring accurate alignment and sharper images. |
| Image Processing Algorithms | Advanced methods, such as Fourier interpolation and particle polishing, refine image quality by reducing noise and correcting distortions, leading to more reliable 3D reconstructions. |
These advancements in technology and processing techniques help address common cryo-EM challenges, improving both the quality and efficiency of structural analysis.
Case Study: High-Resolution Cryo-EM Data Processing of the PaaZ Enzyme
This case study examines the data processing workflow in single-particle cryo-EM, highlighting key computational steps and their impact on the final resolution and quality of reconstructed structures. The PaaZ enzyme case demonstrates the effectiveness of these methods, achieving a 2.9 Å resolution and providing detailed structural and dynamic insights.
Data Acquisition and Preprocessing
In a recent study, 502 micrographs were collected using a Titan Krios microscope equipped with a direct electron detector. Each micrograph contained approximately 220 particles of the bacterial enzyme PaaZ, resulting in a total of 118,000 particle images. The use of DEDs, which can detect single electrons and operate at high frame rates, has significantly improved the signal-to-noise ratio and resolution of cryo-EM structures.
The first step in preprocessing is motion correction, which addresses beam-induced motion during imaging. In this study, motion correction was performed using the MotionCor2 software, which aligns and averages individual frames of the micrographs. This step reduced the B-factor from an initial value of 150 Ų to approximately 100 Ų, indicating a significant improvement in image quality.
CTF Estimation and Correction
The CTF parameters, including defocus and astigmatism, were estimated using the CTFFIND4 software. The defocus values ranged from 1.5 to 3.0 µm, with an average astigmatism of 50 nm. CTF correction was performed using phase flipping, which corrects the phase of the CTF but not its amplitude. This step was essential for accurately reconstructing the 3D structure, as uncorrected CTF distortions can lead to significant errors in the final model.
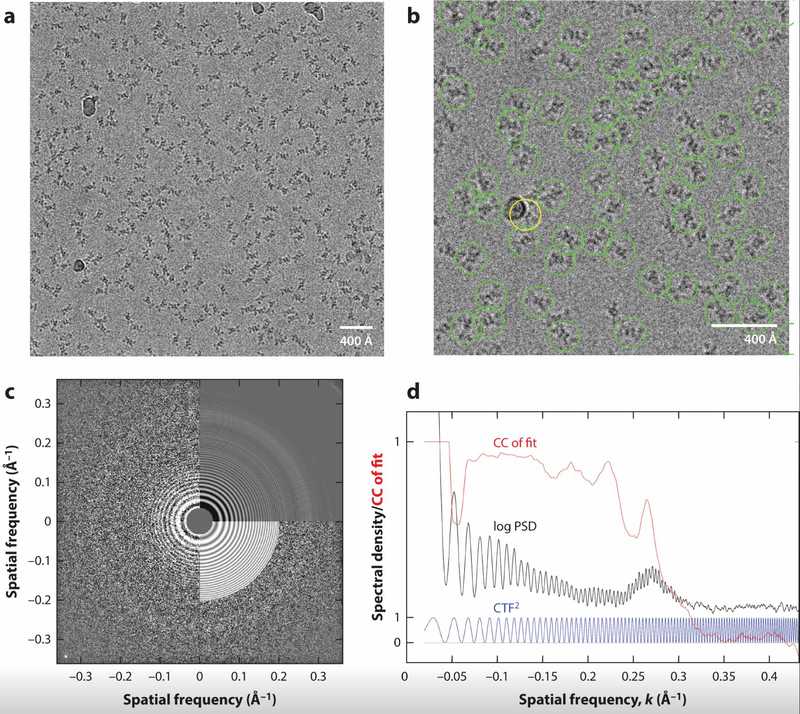 Figure 1. Cryo-EM Micrograph of PaaZ Protein Particles. (a) Overview of a cryo-EM micrograph showing a 434 × 434 nm area containing around 220 PaaZ particles, with some contaminating ice balls. (b) Close-up of selected PaaZ particles using template matching, with an error in one selection (yellow circle) due to an ice ball. (c) CTF fitting to the 2D power spectrum of the micrograph, showing defocus and astigmatism values. (d) 1D power spectral density (PSD) curve and its comparison to the theoretical CTF, highlighting the contribution from the amorphous ice. (Singer A, et al., 2020)
Figure 1. Cryo-EM Micrograph of PaaZ Protein Particles. (a) Overview of a cryo-EM micrograph showing a 434 × 434 nm area containing around 220 PaaZ particles, with some contaminating ice balls. (b) Close-up of selected PaaZ particles using template matching, with an error in one selection (yellow circle) due to an ice ball. (c) CTF fitting to the 2D power spectrum of the micrograph, showing defocus and astigmatism values. (d) 1D power spectral density (PSD) curve and its comparison to the theoretical CTF, highlighting the contribution from the amorphous ice. (Singer A, et al., 2020)
 Figure 2. Defocus Contrast in Cryo-EM Image Formation. (a) Projection of the experimental density map of the PaaZ enzyme. (b) Projection of the map at the optimal orientation from a particle image. (c) Filtered projection using the contrast transfer function, showing defocus-induced information delocalization. (d) Experimental image of the particle, showing the large image size needed to capture delocalized information. (Singer A, et al., 2020)
Figure 2. Defocus Contrast in Cryo-EM Image Formation. (a) Projection of the experimental density map of the PaaZ enzyme. (b) Projection of the map at the optimal orientation from a particle image. (c) Filtered projection using the contrast transfer function, showing defocus-induced information delocalization. (d) Experimental image of the particle, showing the large image size needed to capture delocalized information. (Singer A, et al., 2020)
Particle Picking and 2D Classification
Particle picking is the process of identifying and extracting individual particle images from the micrographs. In this study, a template-matching approach was used, where low-resolution templates generated from negative staining were cross-correlated with the micrographs. This method identified 86,400 high-quality particles out of the initial 118,000, with a false-positive rate of less than 5%.
Following particle picking, 2D classification was performed to group particles with similar views. The RELION software was used for this step, resulting in 50 distinct 2D class averages. These class averages provided an initial assessment of the data quality and were used to remove outliers and poorly aligned particles. The 2D classification step improved the overall SNR by a factor of 2, as measured by the correlation coefficient between the class averages and the raw particle images.
 Figure 3. Particle Images and 2D Class Averages in Cryo-EM. (Left) A gallery of particle images extracted from micrographs, cropped and contrast-reversed. (Right) 2D class averages derived from 118,000 particles using ML-EM in Relion 2.0, with percentages showing the contribution of each class. (Singer A, et al., 2020)
Figure 3. Particle Images and 2D Class Averages in Cryo-EM. (Left) A gallery of particle images extracted from micrographs, cropped and contrast-reversed. (Right) 2D class averages derived from 118,000 particles using ML-EM in Relion 2.0, with percentages showing the contribution of each class. (Singer A, et al., 2020)
3D Reconstruction and Refinement
The 3D reconstruction process began with an initial model generated using the stochastic gradient descent (SGD) algorithm. This model was refined using the maximum likelihood estimation (MLE) approach implemented in RELION. The refinement process involved iterative alignment and averaging of the particle images, with each iteration improving the resolution of the 3D map.
The final 3D reconstruction achieved a resolution of 2.9 Å, as determined by the Fourier shell correlation (FSC) at a threshold of 0.143. The high resolution allowed for the construction of an atomic model of the PaaZ enzyme, revealing detailed structural features such as alpha helices and beta sheets. The model was validated using MolProbity, which reported a clash score of 2.5 and a Ramachandran plot with 98% of residues in favored regions.
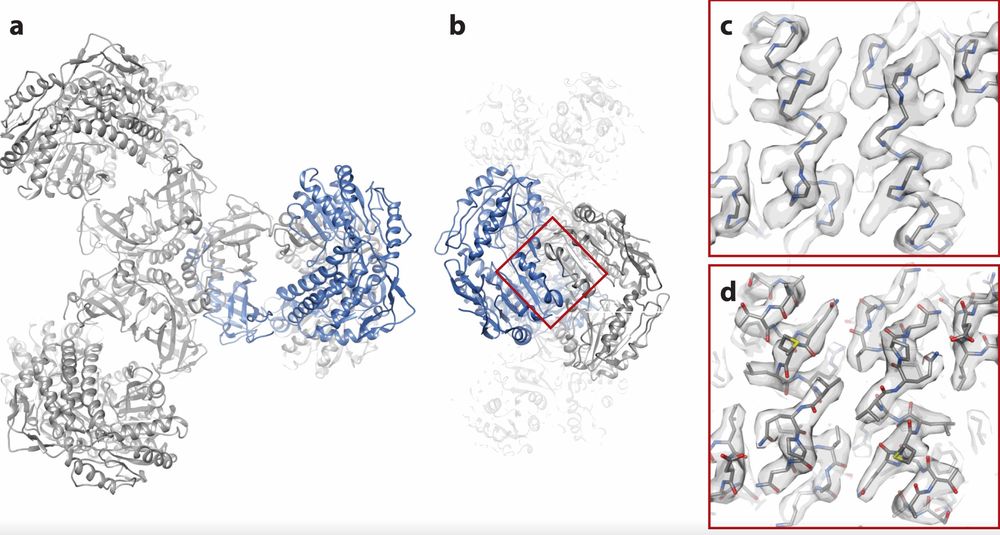 Figure 4. Fitting an Atomic Model to the PaaZ Reconstruction. (a) Overview of the PaaZ model with polypeptide chains and one subunit in blue. (b) End view of the model after a 90° rotation. (c) Superposition of the polypeptide backbone on the density map for two alpha helices. (d) Comparison of the map and atomic model with non-hydrogen atoms shown. (Singer A, et al., 2020)
Figure 4. Fitting an Atomic Model to the PaaZ Reconstruction. (a) Overview of the PaaZ model with polypeptide chains and one subunit in blue. (b) End view of the model after a 90° rotation. (c) Superposition of the polypeptide backbone on the density map for two alpha helices. (d) Comparison of the map and atomic model with non-hydrogen atoms shown. (Singer A, et al., 2020)
Heterogeneity Analysis
One of the advantages of cryo-EM is its ability to analyze structural heterogeneity. In this study, 3D classification was performed to identify different conformational states of the PaaZ enzyme. The classification revealed three distinct states, with populations of 60%, 30%, and 10%, respectively. The major state corresponded to the fully active enzyme, while the minor states represented partially inactive conformations. This analysis provided insights into the functional dynamics of the enzyme and its mechanism of action.
In conclusion, the data processing pipeline in cryo-EM single-particle analysis is a complex but essential process that transforms raw micrographs into high-resolution 3D structures. Key steps such as motion correction, CTF estimation, particle picking, 2D classification, 3D reconstruction, and heterogeneity analysis each contribute to the final resolution and quality of the reconstructed model.
At Creative Biostructure, we offer comprehensive single-particle cryo-EM services, including expert data processing support. Whether you're looking for expert support in structural data processing and interpretation or need assistance with cryo-EM data analysis, we're here to help. Contact us today to discuss your project requirements and discover how our cryo-EM expertise can accelerate your research.
References
- Punjani A. Single-particle CryoEM: Data Processing Techniques for Obtaining Optimal Results. Microscopy and Microanalysis. 2018, 24(S1): 2328-2329.
- Sathyanarayanan N, Cannone G, Gakhar L, et al. Molecular basis for metabolite channeling in a ring opening enzyme of the phenylacetate degradation pathway. Nature Communications. 2019, 10(1): 4127.
- Singer A, Sigworth F J. Computational methods for single-particle electron cryomicroscopy. Annual Review of Biomedical Data Science. 2020, 3(1): 163-190.
- Yan Y, Fan S, Yuan F, et al. A comprehensive foundation model for cryo-EM image processin. bioRxiv, 2024: 2024.11. 04.621604.
- Bhandari, J., Kompaniiets, D., Singh, A.K. et al. Efficient strategies and troubleshooting for single particle cryoEM data collection using EPU. BMC Methods 2, 3 (2025).

-1.jpg)