X-ray crystallography is a cornerstone of structural biology, providing atomic-level insight into macromolecules. This technique has revolutionized drug discovery, enzymology, and biomolecular research by enabling scientists to determine protein structures with high precision. However, obtaining high-resolution structures is not always straightforward. Researchers face many challenges, from crystallization difficulties to data interpretation issues. Understanding and overcoming these common problems is essential to improving the efficiency and success of protein X-ray crystallography.
New to protein X-ray crystallography? Our Beginner's Guide to Protein Crystallography covers the basics.
Issue 1: Challenges in Growing High-Quality Protein Crystals
In protein X-ray crystallography, obtaining high-quality crystals is a crucial step for achieving high-resolution structural determination. However, this process is often hindered by biochemical, physicochemical, and technical challenges. Below, we systematically analyze the key factors affecting crystallization success and discuss practical strategies to overcome them.
Intrinsic Properties of Protein Samples
A. Insufficient Purity and Monodispersity
The purity (>95%) and monodispersity (homogeneity) of a protein sample directly impact crystallization success. Impurities or protein aggregates can disrupt lattice formation, leading to defects or disordered crystals.
Solutions:
- Optimize purification workflows, employing multistep chromatography and carefully designed affinity tags to enhance sample purity. See Optimizing Protein Production and Purification for Crystallography for expert tips.
- Utilize isoelectric focusing (IEF) to remove charge heterogeneity.
- Monitor monodispersity using dynamic light scattering (DLS) to prevent aggregation before crystallization trials.
B. Conformational Dynamics and Surface Properties
Proteins with highly flexible regions (e.g., loops or charged residues) often fail to form stable crystal lattices. For instance, flexible lysine residues on the surface of lysozyme can lead to disordered packing.
Solutions:
- Surface entropy reduction (SER): Replace high-entropy residues (e.g., Lys, Glu) with Ala or Thr to promote crystal contacts.
- Fusion protein strategies: Introduce stable structural domains (e.g., PDZ domains or GST tags) to enhance solubility and facilitate lattice formation.
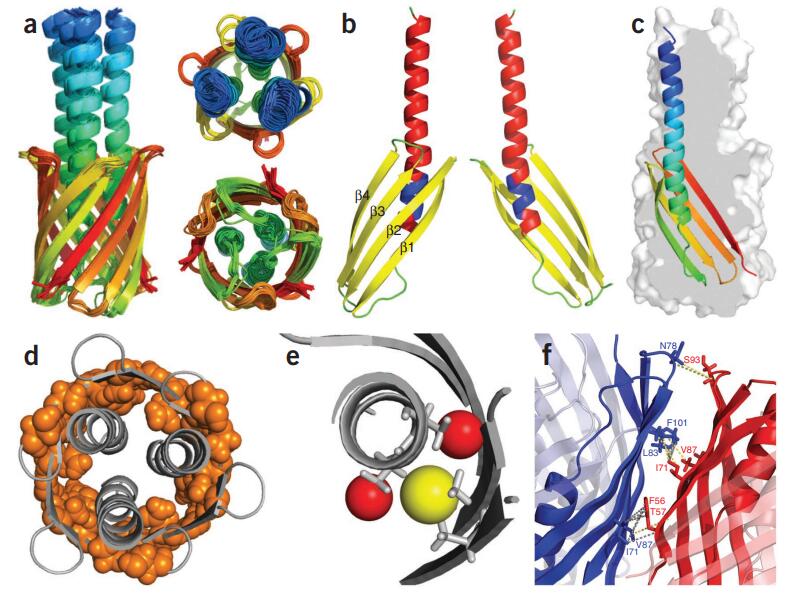
 Figure 1. Protein Fusion Strategies for Membrane Protein Crystallization. Different protein fusion methods for membrane protein crystallization, including terminal fusion, insertion fusion, and termini restraining, with examples from β2 adrenergic receptor and VKOR-like protein structures. (Liu S, et al., 2022)
Figure 1. Protein Fusion Strategies for Membrane Protein Crystallization. Different protein fusion methods for membrane protein crystallization, including terminal fusion, insertion fusion, and termini restraining, with examples from β2 adrenergic receptor and VKOR-like protein structures. (Liu S, et al., 2022)
C. Special Challenges in Membrane Protein Crystallization
Membrane proteins pose additional challenges due to their hydrophobic transmembrane regions, which tend to aggregate and require detergents for solubilization—factors that complicate crystallization.
Solutions:
- Utilize lipidic cubic phase (LCP) or bicelles to mimic the native membrane environment, stabilizing membrane proteins in a crystallization-compatible state.
- Employ fusion with hydrophilic proteins (e.g., T4 lysozyme) to increase solubility and facilitate crystal packing.
Select Service
Related Reading
Complexity of Crystallization Condition Optimization
A. Chemical Condition Screening
Crystallization conditions involve a vast parameter space, including pH, salt concentration, and precipitant type, making trial-and-error methods inefficient. For example, subtle variations in PEG concentration can significantly impact protein solubility.
Solutions:
- Sparse-matrix screening: Utilize historical crystallization data to design a condition library for systematic testing.
- Counter-diffusion method: Gradual mixing of protein and precipitant allows for precise supersaturation control, improving crystallization outcomes.
- Microseed Matrix Screening (MMS): Uses pre-formed microcrystals as nucleation templates to expand crystallization conditions.
B. Influence of Physical Parameters
Environmental factors such as temperature, gravity, and electric fields are often overlooked but significantly impact crystal growth. For instance, temperature fluctuations can shift protein solubility curves, leading to unwanted phase transitions.
Solutions:
- Temperature gradient screening: Evaluate protein stability across a range of 4°C to 37°C to determine optimal crystallization conditions.
- Electric field modulation: Applying a 1 MHz electric field has been shown to improve lysozyme crystal quality by reducing subgrain misalignment.
Technological Innovations in Crystallization
A. Heterogeneous Nucleation for Enhanced Crystal Growth
Traditional homogeneous nucleation relies on high supersaturation, which often results in excessive microcrystal formation. Porous materials such as SDB microspheres or Bioglass can reduce the nucleation energy barrier, promoting controlled crystal growth.
B. Automation and High-Throughput Techniques
Manual crystallization screening is labor-intensive and subject to bias. Automation significantly accelerates condition optimization.
Technological Applications:
- Robotic liquid handling systems (e.g., Crystal Gryphon) enable nanoliter-scale screening, maximizing sample efficiency.
- AI-driven image analysis using convolutional neural networks (CNNs) automates crystal recognition and classification, minimizing human error.
C. Alternative Structure Determination Methods
For proteins that fail to form large single crystals, emerging techniques offer alternative pathways for structural analysis:
- Microcrystal Electron Diffraction (MicroED): Suitable for nanocrystals, providing atomic-resolution structures (e.g., lysozyme microcrystals).
- Small-Angle X-ray Scattering (SAXS): Provides solution-state structural information without the need for crystallization.
Select Service
Post-Crystallization Optimization and Quality Validation
A. Post-Crystallization Treatments to Enhance Diffraction Quality
Even if crystals are obtained, additional steps may be required to improve diffraction resolution.
- Dehydration treatment: Gradually reducing humidity can contract the crystal lattice, improving order and resolution (e.g., dehydrated lysozyme crystals often yield higher-resolution diffraction).
- Ligand soaking: Introducing stabilizing small molecules (e.g., inhibitors) can fill lattice voids, reducing disorder.
B. Quality Assessment Tools for Crystal Evaluation
- MolProbity validation: Identifies stereochemical errors and atomic clashes in refined structures.
- X-ray rocking curve analysis: Measures diffraction peak widths (FWHM) to assess lattice quality and perfection.
Refer to our article From Solution to Crystal: Mastering Protein Crystallization for more details.
Issue 2: Solving the Phase Problem in X-ray Crystallography
What Is Phase Problem in XRD?
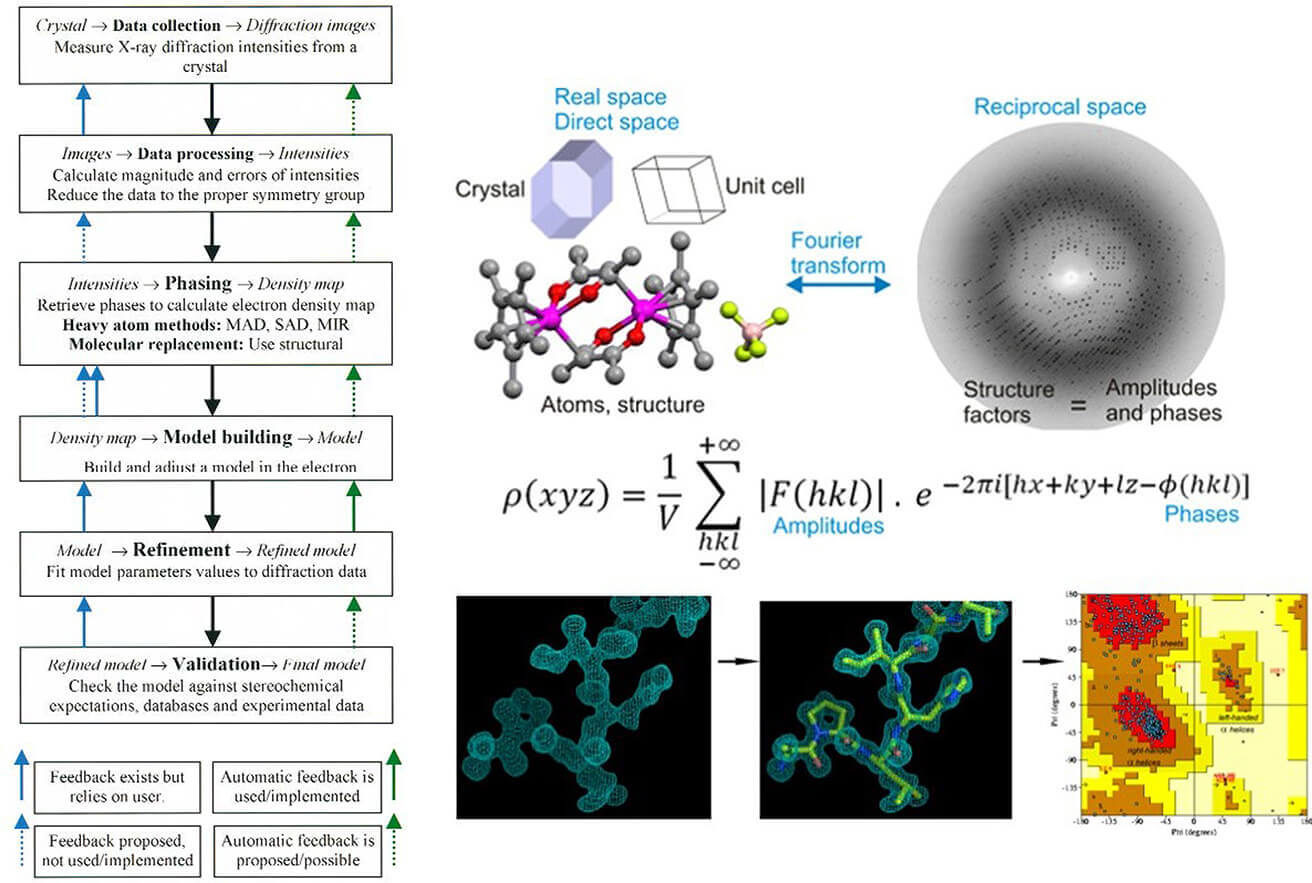
In X-ray crystallography, the phase problem refers to the inability to directly measure the phase information of diffracted X-rays, even though the diffraction pattern provides the amplitude. Since the electron density map—which reveals the 3D atomic structure—requires a Fourier transform incorporating both amplitude and phase, the lack of phase information makes structure determination impossible.
The root of this problem lies in how X-rays interact with crystals. X-ray detectors record only diffraction intensities (which are proportional to the squared amplitude), but phase information is lost because X-ray frequencies (~10¹⁸ Hz) are too high for direct measurement.
Importance of the Phase Problem
The lack of phase information is a major bottleneck in structure determination. The accuracy of electron density maps directly impacts the precise positioning of atoms, affecting our understanding of protein function, such as enzyme active sites or drug-binding pockets.
It is estimated that ~40% of crystallography projects are hindered by the phase problem, especially when working with novel proteins lacking homologous models. This challenge is particularly significant in de novo structure determination where no prior structural data is available.
Major Methods for Solving the Phase Problem
A. Anomalous Scattering Techniques
Anomalous scattering utilizes heavy atoms (e.g., selenium, mercury) that exhibit wavelength-dependent phase shifts, enabling phase determination.
- Single-wavelength anomalous diffraction (SAD) and Multi-wavelength anomalous diffraction (MAD) leverage these shifts to infer phase information.
- Selenium-substituted methionine (Se-Met) labeling has become a dominant method, contributing to over 70% of de novo structures in the PDB.
Technical Challenges:
- Heavy atoms must be successfully incorporated into the crystal, and the diffraction quality must remain high.
- Radiation damage is a major concern with conventional synchrotron sources, as prolonged exposure can degrade crystals.
- X-ray free-electron lasers (XFELs) (e.g., LCLS) mitigate damage through the "diffraction-before-destruction" approach, allowing the use of microcrystals while preserving structural integrity.
 Figure 2. Electron Density Mapping of the Selenobiotin Site. Comparison of electron density at the selenobiotin site before (a) and after (b) refinement. The green density highlights the selenium atom position, aiding accurate structure modeling. (Hunter M S, et al., 2016)
Figure 2. Electron Density Mapping of the Selenobiotin Site. Comparison of electron density at the selenobiotin site before (a) and after (b) refinement. The green density highlights the selenium atom position, aiding accurate structure modeling. (Hunter M S, et al., 2016)
B .Molecular Replacement (MR)
When a homologous structure (sequence identity >30%) is available, molecular replacement (MR) can be used. This method involves rotating and translating a known structure to match the unknown crystal's diffraction pattern, allowing for an initial estimation of phase.
- Integration with machine learning: AlphaFold and RoseTTAFold can generate predicted structures that serve as search models for MR, significantly broadening its applicability. Refer to our article The Evolution of AlphaFold: A Milestone in Protein Structure Prediction for more details.
- Limitations: MR is ineffective for low-homology proteins or highly flexible regions (e.g., transport proteins), where experimental phasing methods are still required.
C. Direct Methods and Density Modification
Direct Methods:
- Based on probabilistic algorithms, direct methods infer phase information from diffraction intensities.
- Effective for small molecules, but large macromolecules lack sufficient data for reliable phase estimation.
Density Modification:
- Iterative phase refinement techniques leverage known physical properties of electron density:
Non-negativity constraint (electron density cannot be negative).
Solvent flattening (density is expected to be uniform in solvent regions).
- Maximum likelihood density modification (MLDM) and solvent flipping have been instrumental in refining low-resolution maps.
- PHENIX AutoBuild integrates density modification and automated model building, accelerating structure refinement.
D. Deep Learning Approaches
- Phase retrieval algorithms:
Hybrid Iterative Optimization (HIO) and Fourier holography (FTH) iteratively refine phase estimates but often get trapped in local minima.
- AI-driven phase recovery:
CrysFormer applies Patterson maps and attention mechanisms to directly infer atomic coordinates from diffraction data.
AlphaFold-integrated crystallography: By incorporating crystallographic data into deep learning models, end-to-end structure prediction without a prior model is now feasible.
Finding our solutions for Crystallography Phase Determination.
Issue 3: Effects and Mitigation Strategies of Radiation Damage
Specific Effects of Radiation Damage
1. Chemical Bond Breakage and Residue Modifications
Radiation exposure leads to the breakage of specific chemical bonds in protein crystals, particularly disulfide bonds (Cys-Cys) and carboxyl groups in acidic residues (Asp, Glu). A notable example is acetylcholinesterase (Torpedo californica), where the catalytic triad residue His-440 is highly susceptible to radiation damage, potentially compromising its enzymatic activity. Active sites and solvent-exposed acidic residues are particularly vulnerable, which can alter ligand binding and catalytic mechanisms.
2. Conformational Bias in Structural Ensembles
While cryogenic temperatures (100 K) reduce overall damage, they can mask dynamic conformations of proteins. Studies suggest that over 35% of side-chain conformations are altered due to freezing, potentially eliminating functionally relevant intermolecular interactions. For example, H-Ras protein crystal structures obtained at room temperature (RT) revealed an allosteric network consistent with NMR findings, whereas cryogenic data failed to capture these dynamics.
3. Degradation of Diffraction Quality
Radiation damage causes progressive intensity decay in diffraction patterns, with an estimated half-dose threshold of ~4.3×10⁷ Gy. When exceeding a critical dose (~1×10⁷ Gy), secondary damage (e.g., free radical diffusion) accelerates, leading to crystal amorphization and loss of diffraction power.
4. Overfitting and Structural Model Bias
Cryogenic data tends to produce overly compact, static models, potentially masking structural heterogeneity required for catalysis and allosteric regulation. Such biases contribute to misrepresentations in structural databases, limiting accurate functional interpretations.
Technical Approaches to Mitigate Radiation Damage
A. Cryogenic Cooling
Principle: Cooling to 100 K (liquid nitrogen) reduces free radical migration, slowing secondary radiation damage.
Application Example: Ferritin (apoferritin/holoferritin) crystals withstand radiation doses up to 3.0×10⁷ Gy at 100 K, with controlled data degradation.
Limitations: Freezing artifacts may obscure functionally important conformations, requiring room-temperature validation.
 Figure 3. Impact of Cryocooling on Protein Crystal Structure and Packing. (A) Comparative analysis shows cryocooling causes greater reduction in unit-cell volume than in protein volume, likely due to solvent expulsion. (B) Packing defects in CypA at room temperature (red) and cryogenic conditions (blue) highlight both shared and temperature-specific structural variations, including functionally relevant alternative conformations. (Fraser J S, et al., 2011)
Figure 3. Impact of Cryocooling on Protein Crystal Structure and Packing. (A) Comparative analysis shows cryocooling causes greater reduction in unit-cell volume than in protein volume, likely due to solvent expulsion. (B) Packing defects in CypA at room temperature (red) and cryogenic conditions (blue) highlight both shared and temperature-specific structural variations, including functionally relevant alternative conformations. (Fraser J S, et al., 2011)
B. X-ray Parameter Optimization
High-energy, short-wavelength X-rays: Using X-rays beyond Cu Kα reduces photon absorption, lowering per-unit-dose damage.
Dose-limiting strategies: Tools provide real-time monitoring of accumulated dose, ensuring exposure remains within recommended limits (~3.0×10⁷ Gy).
C. Rapid Data Collection Techniques
Synchrotron-based Serial Crystallography (SSX): Room-temperature scanning of multiple microcrystals using millisecond-scale exposures prevents excessive dose accumulation.
X-ray Free Electron Lasers (XFELs): Femtosecond pulses (<50 fs) collect diffraction data before damage occurs. LCLS experiments collected 2.5 Å resolution data from lysozyme microcrystals at 10 Hz, reducing sample consumption by 90%.
Protein X-ray crystallography is a powerful technique for determining macromolecular structures, but challenges like crystallization difficulties, phase determination issues, and radiation damage can hinder success. By applying advanced strategies and emerging technologies, researchers can significantly improve data quality and resolution. At Creative Biostructure, we offer comprehensive protein crystallography services, from sample preparation to high-resolution structure determination. Contact us to optimize your crystallography workflow and accelerate your structural biology research.
References
- Fraser J S, Van Den Bedem H, Samelson A J, et al. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proceedings of the National Academy of Sciences. 2011, 108(39): 16247-16252. https://doi.org/10.1073/pnas.1111325108
- Guo Y Z, Sun L H, Oberthuer D, et al. Utilisation of adsorption and desorption for simultaneously improving protein crystallisation success rate and crystal quality. Scientific Reports. 2014, 4(1): 7308. https://doi.org/10.1038/srep07308
- Hunter M S, Yoon C H, DeMirci H, et al. Selenium single-wavelength anomalous diffraction de novo phasing using an X-ray-free electron laser. Nature Communications. 2016, 7(1): 13388. https://doi.org/10.1038/ncomms13388
- Holcomb J, Spellmon N, Zhang Y, et al. Protein crystallization: Eluding the bottleneck of X-ray crystallography. AIMS Biophysics. 2017, 4(4): 557. https://doi.org/10.3934/biophy.2017.4.557
- Liu S, Li W. Protein fusion strategies for membrane protein stabilization and crystal structure determination. Crystals. 2022, 12(8): 1041. https://doi.org/10.3390/cryst12081041