Protein X-ray crystallography has been a cornerstone of drug discovery for decades, providing atomic-level insights into protein structures. This technique allows researchers to determine how small molecules interact with biological targets, enabling the rational design of new therapeutics. In this article, we explore the principles of X-ray crystallography, its role in drug development, and the latest advancements improving its application in modern pharmaceutical research.
What is Protein X-ray Crystallography and How Does It Work?
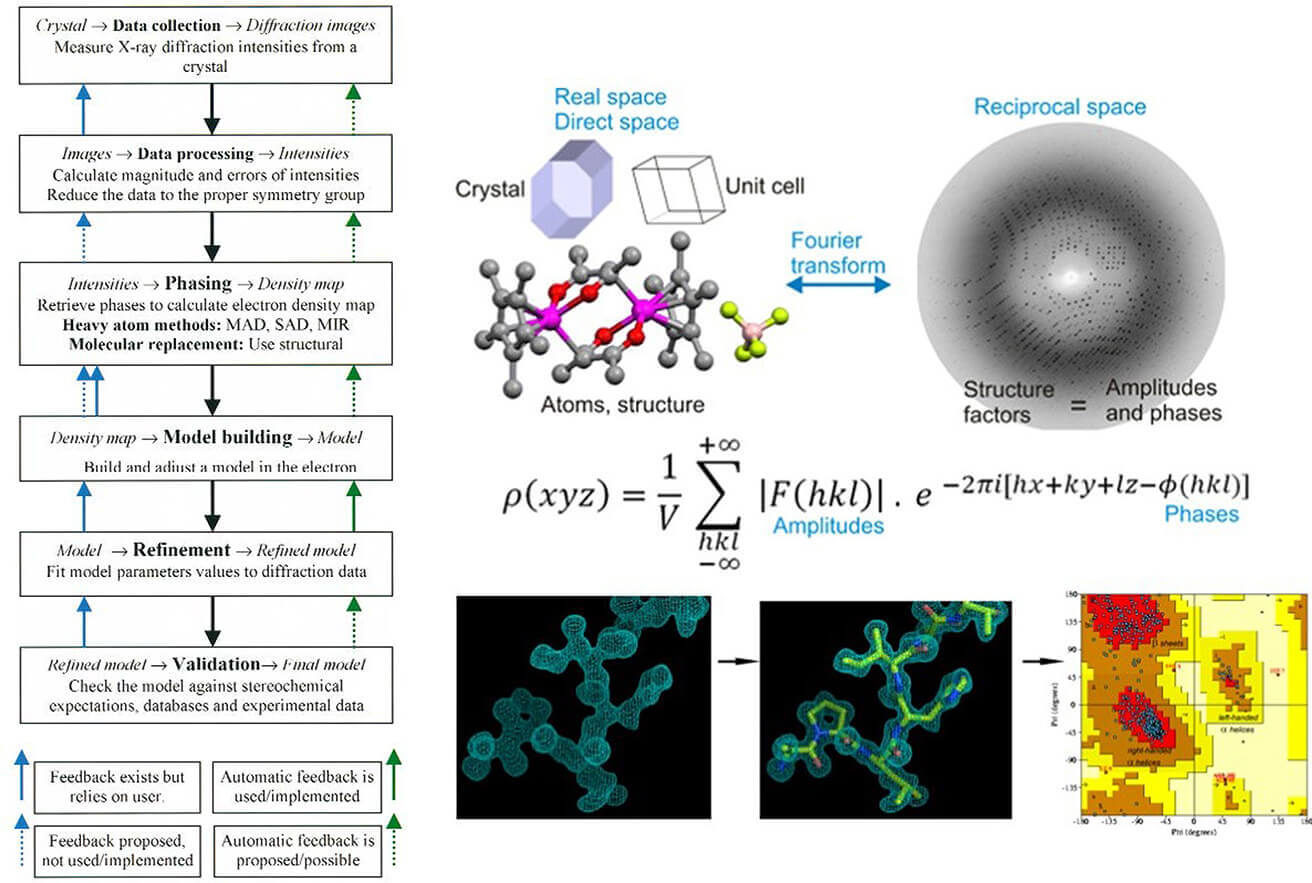
X-ray crystallography is a structural determination method that relies on the diffraction of X-rays through a protein crystal. The technique follows these key steps:
- Protein Crystallization: The target protein is purified and crystallized under controlled conditions. This step is crucial, as well-formed crystals produce high-quality diffraction patterns.
- X-ray Diffraction: The crystal is exposed to X-rays, generating a diffraction pattern that represents the electron density of the protein.
- Data Processing and Structure Determination: Computational algorithms reconstruct the 3D atomic structure of the protein from the diffraction data.
- Model Refinement and Interpretation: The structure is refined for accuracy and analyzed to understand biological functions or interactions with small molecules.
Refer to our article A Beginner's Guide to Protein Crystallography for more details.
Role of Protein X-ray Crystallography in Drug Discovery
X-ray crystallography plays a core role in drug discovery. Its high-resolution structural analysis capability provides atomic-level information support for target identification, drug design and optimization. The following are its specific applications in various links:
A. Identifying Drug Targets
X-ray crystallography plays a crucial role in drug discovery by resolving the three-dimensional structures of potential drug targets, allowing researchers to analyze their functional sites and binding characteristics. This structural insight is essential for understanding protein function and guiding the design of targeted therapeutics.
Structural Determination of Therapeutic Targets
X-ray crystallography enables the identification of key functional sites in proteins, such as active sites, allosteric sites, and protein-nucleic acid interaction interfaces. These structural details provide the foundation for rational drug design.
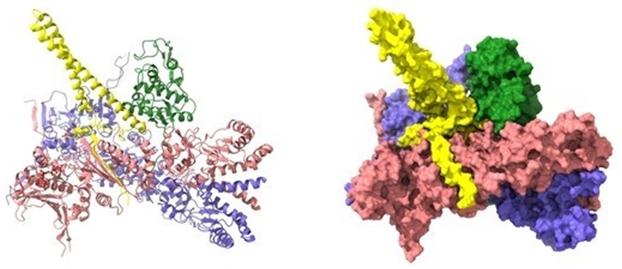
One key example is in Alzheimer's disease research, where X-ray crystallography helped elucidate the high-resolution structures of β-amyloid (Aβ) oligomers—key contributors to amyloid plaque formation. Structures such as the Aβ trimer (PDB: 5SUT) and dodecamer (PDB: 5SUR) provided a foundational understanding of Aβ aggregation, facilitating the development of drugs aimed at inhibiting amyloid deposition.
 Figure 1. X-ray crystallographic structures of β-amyloid (Aβ) oligomers. (Kreutzer A G, et al., 2018)
Figure 1. X-ray crystallographic structures of β-amyloid (Aβ) oligomers. (Kreutzer A G, et al., 2018)
Discovery of Cryptic Binding Sites
Cryptic binding sites are transient or hidden pockets that remain undetectable in ligand-free protein structures but become accessible under certain physiological conditions. X-ray crystallography plays a key role in identifying such sites through ligand-induced conformational changes and solvent-assisted simulations.
- Capturing Dynamic Conformations
By integrating molecular dynamics simulations with X-ray crystallographic data, researchers can confirm ligand-induced structural shifts that expose cryptic sites. A notable example is the SARS-CoV-2 spike protein, where the cryptic epitope for the CR3022 antibody is only accessible when the receptor-binding domain (RBD) adopts an "open" conformation. This finding, derived from crystallographic and computational studies, provided new targets for therapeutic antibody development.
- Solvent-Based Binding Site Prediction
Another approach involves mixed-solvent molecular dynamics (MixMD) simulations, which model how proteins interact in organic solvent-water mixtures to predict hidden pockets. X-ray crystallography then validates these cryptic sites by determining their geometry and ligand-binding potential, enhancing drug design strategies.
Target Validation and Druggability Assessment
Determining whether a biological target is druggable—meaning it possesses favorable characteristics for small-molecule binding—is a critical step in early-stage drug discovery. X-ray crystallography supports this assessment by analyzing binding pocket properties and conformational flexibility.
- Binding Site Characterization
Crystallographic data allow researchers to quantify key parameters such as pocket volume, hydrophobicity, and hydrogen-bond networks, which influence druggability. For example, computational tools like CAVITY analyze protein surface grooves in crystal structures to predict the likelihood of small-molecule binding.
- Conformational Druggability Analysis
Some drug targets exhibit pH-dependent conformational shifts that affect ligand binding. The influenza M2 proton channel, for instance, undergoes structural rearrangements at low pH, directly influencing the binding efficiency of inhibitors like amantadine. This pH-driven druggability assessment was achieved through X-ray crystallography combined with NMR studies, providing insights for optimizing antiviral drugs.
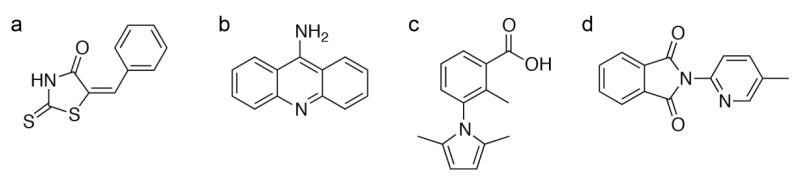
 Figure 2. Binding Modes of Inhibitors at the Active Site of Human Chymase. (Arooj M, et al., 2013)
Figure 2. Binding Modes of Inhibitors at the Active Site of Human Chymase. (Arooj M, et al., 2013)
B. Structure-Based Drug Design (SBDD)
X-ray crystallography plays a central role in drug discovery, especially in Structure-Based Drug Design (SBDD). It provides key structural information for virtual screening, lead optimization, and binding mode analysis by analyzing the high-resolution three-dimensional structure of the target protein and its ligand complex. The following is a detailed analysis of specific applications and mechanisms:
Virtual Screening
Virtual screening utilizes computational approaches to identify potential bioactive molecules from large compound libraries. X-ray crystallography plays a crucial role in this process by providing high-resolution structural data for accurate molecular modeling.
- Target Structure Modeling
The X-ray-resolved structures of target proteins, such as enzymes and receptors, serve as the foundation for virtual screening. These structures enable the generation of pharmacophore models and docking simulations, aiding in the identification of promising drug candidates. For instance, X-ray crystal structures of HIV protease and angiotensin-converting enzyme (ACE) have been extensively used for virtual screening and structure-based drug discovery.
- Validation of Docking Accuracy
After virtual screening, the predicted ligand conformations need to be verified against experimentally determined X-ray structures. Comparing computational docking results with crystal structures of protein-ligand complexes ensures the reliability of molecular docking algorithms. For example, in a study on human chymase inhibitors, docking predictions were cross-referenced with X-ray structures to confirm binding accuracy.
Lead Optimization
X-ray crystallography provides atomic-level interaction details that are essential for refining lead compounds to improve potency, selectivity, and pharmacokinetic properties.
- Binding Site Analysis
By resolving the complex structures of lead compounds with target proteins, researchers can identify key interactions such as hydrogen bonds, hydrophobic contacts, and π-π stacking interactions. These insights guide rational chemical modifications to enhance binding affinity. For example, multiple rounds of X-ray structural refinement have been employed in the design of HIV protease inhibitors, enabling the stepwise optimization of side chains for improved efficacy.
- Capturing Dynamic Conformations
Some drug targets undergo ligand-induced conformational changes upon binding, a phenomenon known as induced fit. X-ray crystallography captures these dynamic structural shifts, helping optimize drug interactions. For instance, p38 MAP kinase exhibits an active site conformational adjustment upon fragment binding, which was revealed through X-ray analysis, providing insights for further compound optimization.
- Multi-Target Selectivity Optimization
For drug development efforts requiring selectivity between protein subtypes, X-ray crystallography enables comparative structural analysis. By examining subtle structural differences among closely related targets, researchers can design selective inhibitors. A notable example is the development of subtype-selective estrogen receptor (ERα/β) ligands, where X-ray structures of different receptor subtypes guided the design of highly selective compounds.
Binding Mode Analysis
X-ray crystallography provides direct visualization of drug-target interactions, ensuring accurate modeling of binding modes.
- High-Resolution Electron Density Mapping
High-resolution electron density maps reveal the precise orientation and conformational isomers of bound ligands, reducing the risk of incorrect docking predictions. For example, the SARS-CoV-2 Nsp1 protein fragment binding site was elucidated through X-ray crystallography, ensuring accurate placement of drug candidates in computational models.
- Allosteric Modulation Mechanisms
X-ray crystallography also uncovers allosteric binding sites and long-range conformational changes induced by ligand binding. This is particularly valuable for designing allosteric inhibitors. One example is the integrin receptor, where X-ray structures revealed a ligand-induced allosteric inhibition mechanism, crucial for developing novel anti-inflammatory therapeutics.
- Validation of Covalent Inhibitor Binding
For covalent inhibitors, X-ray crystallography is indispensable in confirming covalent bond formation at the binding site and elucidating reaction mechanisms. This approach has been widely applied in kinase inhibitor development, ensuring accurate characterization of covalent drug-target interactions.
C. Fragment-Based Drug Discovery and X-ray Crystallography
High-Throughput Fragment Screening and Structural Analysis
X-ray crystallography is a key tool in FBDD, helping researchers analyze small molecular fragments (typically <300 Da) bound to target proteins. Even though these fragments have weak binding affinities (Kd values in the μM to mM range), X-ray crystallography can reveal their binding modes with atomic-level detail.
Unlike high-throughput screening (HTS), which tests hundreds of thousands of compounds, fragment libraries are much smaller (500-2000 molecules), allowing for systematic structural analysis. Using co-crystal structures, researchers can determine how fragments bind, identify key hydrogen bonds and hydrophobic interactions, and guide fragment optimization. This supports fragment growing (adding new chemical groups to improve binding) and fragment linking (connecting fragments that bind close together).
Binding Mode-Guided Optimization
X-ray crystallography helps optimize fragments by providing structural insights into their binding. Key strategies include:
- Enhancing hydrogen bonding: Strengthening interactions with key protein residues to improve binding.
- Exploiting conformational changes: Some fragments induce shape changes in proteins, which can be used to design allosteric inhibitors.
- Replacing water molecules: Identifying and replacing high-energy water molecules in the binding pocket to improve stability.
Advantages and Challenges
Advantages
- Provides high-resolution (often <1.5 Å) structural details.
- Detects weakly binding fragments, even when interactions are unstable.
Despite these challenges, X-ray crystallography remains a powerful method in FBDD, offering critical insights for drug design and optimization.
D. Understanding Drug Resistance
Drug resistance often arises from mutations in target proteins. X-ray crystallography helps identify structural changes caused by mutations, guiding the development of next-generation inhibitors that overcome resistance.
Structural Basis of Drug Resistance
X-ray crystallography helps uncover drug resistance mechanisms by comparing wild-type and mutant protein structures. Key resistance mechanisms include:
- Direct steric hindrance: Some mutations create spatial clashes that prevent drug binding. For example, the V82I mutation in HIV protease introduces a bulky side chain, interfering with inhibitor binding.
- Allosteric conformational changes: Mutations can alter the active site shape, reducing drug affinity. For instance, the Q80K mutation in HCV NS3/4A protease shifts the enzyme's conformation, weakening inhibitor interactions.
- Binding pocket remodeling: Certain mutations expand or reshape the binding site, reducing drug efficacy. The T790M mutation in EGFR kinase enlarges the ATP-binding pocket, making it harder for inhibitors to bind effectively.
Strategies to Overcome Drug Resistance
Structural insights from X-ray crystallography guide the development of next-generation drugs that counteract resistance:
- Dual-target inhibitors: Some drugs are designed to bind both wild-type and mutant proteins. For example, osimertinib, a third-generation EGFR inhibitor, effectively targets both normal and resistant EGFR variants.
- Allosteric inhibitors: By binding to alternative sites instead of the drug-resistant active site, these inhibitors bypass common resistance mutations. For instance, asciminib, an allosteric ABL1 kinase inhibitor, binds away from the ATP site, avoiding mutations that confer resistance to traditional kinase inhibitors.
E. Validating Drug Candidates
Once a lead compound is developed, crystallography provides structural validation, ensuring that the compound binds as intended. This is essential for optimizing pharmacodynamics and pharmacokinetics.
Binding Mode Verification
X-ray crystallography serves as a gold-standard method for confirming drug binding, ensuring:
- Ligand orientation accuracy: Distinguishing stereoisomer binding differences, such as in HIV protease inhibitors, where enantiomers may exhibit varying affinities.
- Docking model validation: Computational docking predictions must be experimentally verified, especially for flexible ligands with multiple rotatable bonds.
Comprehensive Structural Validation
Multiple structural parameters must be assessed to ensure binding accuracy:
- Electron density quality: The 2mF₀−DFc electron density map must be well-defined around the ligand to prevent overfitting—a known issue in ~15% of PDB small-molecule structures.
- B-factor and occupancy analysis: High B-factors or low occupancy indicate binding instability, suggesting the need for ligand rigidity optimization.
Integrating Multiple Validation Techniques
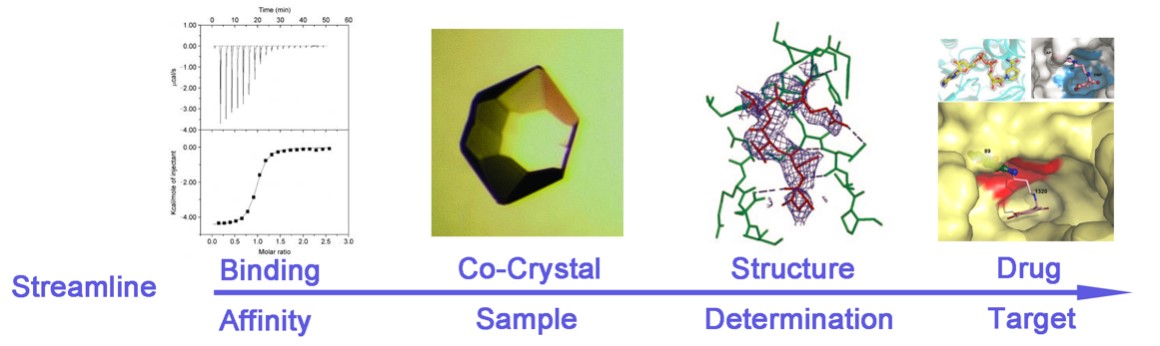
X-ray crystallography is most effective when integrated with complementary biophysical methods for comprehensive drug evaluation. Surface plasmon resonance (SPR) provides kinetic insights into binding interactions, while isothermal titration calorimetry (ITC) reveals thermodynamic parameters such as binding affinity and enthalpy changes. Additionally, co-crystal structures offer direct visualization of ligand binding, ensuring precise validation of drug candidates.
 Figure 3. The Drug Design Cycle and Role of X-ray Crystallography. (Maveyraud L, et al., 2020)
Figure 3. The Drug Design Cycle and Role of X-ray Crystallography. (Maveyraud L, et al., 2020)
Advantages of X ray Crystallography in Drug Design
Atomic-Level Resolution for Structural Analysis
X-ray crystallography provides high-resolution (up to 1.5 Å) three-dimensional structures of proteins and their complexes, enabling precise visualization of ligand binding sites, hydrogen bond networks, and hydrophobic interactions. This atomic-level detail is essential for understanding molecular recognition and guiding SBDD. Moreover, high-resolution data allow differentiation between stereoisomers and conformers, ensuring accurate drug design.
Empowering Structure-Based Drug Design (SBDD)
- Target Validation: By resolving the structures of disease-related proteins (e.g., kinases, GPCRs, and viral enzymes), X-ray crystallography helps confirm the feasibility of these targets for drug development.
- Ligand Optimization: Structural insights from co-crystal complexes provide guidance for modifying lead compounds to enhance binding affinity and selectivity. For example, binding mode analysis can reveal key interactions with active-site residues, aiding in lead optimization.
- FBDD: Even for weak-affinity fragments (Kd in the μM to mM range), X-ray crystallography can determine precise binding orientations, accelerating the transition from fragment hits to optimized drug candidates.
Broad Applicability Across Biomolecular Systems
Beyond soluble proteins, X-ray crystallography has successfully elucidated the structures of viral capsids, immune complexes, and protein-nucleic acid assemblies. Comparative analyses between ligand-bound and unbound structures provide insights into conformational changes and receptor activation mechanisms.
Well-Established Computational and Database Support
Software suites like CCP4, Phenix, and XDS streamline every step of data processing, from phasing to model refinement, improving workflow efficiency. Additionally, over 90% of Protein Data Bank (PDB) entries originate from X-ray crystallography, offering a vast structural reference for drug discovery projects.
Limitations of X-ray Crystallography in Drug Discovery
Challenges in Sample Preparation
- Crystallization Bottlenecks: Many biologically relevant proteins, particularly membrane proteins (e.g., ion channels, GPCRs) and intrinsically disordered proteins, are difficult to crystallize. This challenge can extend structure determination timelines to several years.
- High-Quality Crystal Requirements: Large, defect-free crystals (often in the micrometer range) are necessary for high-resolution diffraction. Small or radiation-sensitive crystals require synchrotron or XFEL-based approaches to mitigate damage.
Static Nature of Crystal Structures
- Limited Ability to Capture Dynamic States: Traditional X-ray crystallography provides only a static "snapshot" of a protein's lowest-energy conformation, making it challenging to study enzyme catalysis, transient binding states, or allosteric transitions.
- Invisible Hydrogen Atoms: Since hydrogen atoms scatter X-rays weakly, key interactions such as proton transfer in enzymatic reactions often require additional neutron diffraction or computational modeling.
Data Processing and Interpretation Challenges
- Phase Problem: Structure determination relies on molecular replacement or heavy-atom derivatization, which can introduce errors if no homologous reference structures exist.
- Modeling Bias: Incorrect interpretation of electron density maps may lead to misoriented side chains or incorrect ligand placement, necessitating careful refinement with validation tools like MolProbity.
Competition with Emerging Structural Techniques
Advances in single-particle cryo-electron microscopy (cryo-EM) have enabled structure determination at 2-4 Å resolution without crystallization, making it the preferred method for large protein complexes, membrane proteins, and flexible biomolecules. While time-resolved serial crystallography (TR-SFX) enables kinetic studies, cryo-EM can directly capture multiple conformational states within a single dataset, offering a more comprehensive view of molecular motions.
Refer to our article Single Particle Cryo-EM Overview for more details.
The Future of X-ray Crystallography in Drug Discovery
Integration with Emerging Technologies
X-ray crystallography is becoming increasingly powerful when combined with other structural biology techniques:
- Synergy with Cryo-EM: Cryo-EM provides structural data on large macromolecular complexes in their native and dynamic states, while X-ray crystallography offers atomic-level resolution of static structures. Hybrid models that integrate both methods enhance structural accuracy, particularly for challenging targets like membrane proteins and viral particles.
- AI-Driven Structural Modeling: AI tools like AlphaFold and RoseTTAFold are transforming drug discovery by predicting protein structures with high accuracy, accelerating SBDD. AI-powered molecular dynamics simulations further refine drug-target interactions, while graph neural networks (GNNs) enhance fragment-based drug design. Refer to our article Integrating AlphaFold into the Drug Discovery Process for more details.
- Multi-Technique Integration: Advanced approaches combine mass spectrometry (MS) for fragment-based screening, NMR spectroscopy for studying protein flexibility, and X-ray crystallography for high-resolution validation. This integrative approach allows a more comprehensive understanding of biomolecular interactions.
Technological Advancements Enhancing Efficiency
Several innovations are making X-ray crystallography faster and more efficient:
- Automation and High-Throughput Processing: Robotic systems like JBluIce-EPICS automate crystal localization and diffraction data collection, increasing throughput by over tenfold. Deep learning models, such as convolutional neural networks (CNNs), classify crystallographic images with over 95% accuracy, reducing human intervention.
- MicroED for Nano-Crystals: MicroED enables structure determination from nanometer-sized crystals, overcoming limitations of traditional crystallography. This method provides high-resolution data in under an hour, making it ideal for studying drug polymorphs and small-molecule structures.
- Next-Generation Synchrotrons and XFEL: Facilities like APS-U and XFEL are revolutionizing time-resolved crystallography, capturing enzyme catalysis and transient intermediate states with unprecedented accuracy. These breakthroughs are critical for mechanism-driven drug design.
With continuous advancements and interdisciplinary integration, X-ray crystallography remains an indispensable tool in drug discovery, evolving to meet the challenges of modern pharmaceutical research.
At Creative Biostructure, we offer expert X-ray crystallography services to support your drug discovery efforts. Contact us today to learn how we can help accelerate your research and bring your drug candidates to life.
References
- Arooj M, Kim S, Sakkiah S, et al. Molecular modeling study for inhibition mechanism of human chymase and its application in inhibitor design. PLoS One. 2013, 8(4): e62740. https://doi.org/10.1371/journal.pone.0062740
- Kreutzer A G, Nowick J S. Elucidating the structures of amyloid oligomers with macrocyclic β-hairpin peptides: Insights into Alzheimer's disease and other amyloid diseases. Accounts of chemical research. 2018, 51(3): 706-718. https://doi.org/10.1021/acs.accounts.7b00554
- Bancet A, Raingeval C, Lomberget T, et al. Fragment linking strategies for structure-based drug design. Journal of Medicinal Chemistry. 2020, 63(20): 11420-11435. https://doi.org/10.1021/acs.jmedchem.0c00242
- Maveyraud L, Mourey L. Protein X-ray crystallography and drug discovery. Molecules. 2020, 25(5): 1030. https://doi.org/10.3390/molecules25051030
- Narasimhan S. Determining protein structures using X-Ray crystallography. Plant Functional Genomics: Methods and Protocols, Volume 1. New York, NY: Springer US, 2024: 333-353. https://doi.org/10.1007/978-1-0716-3778-4_23