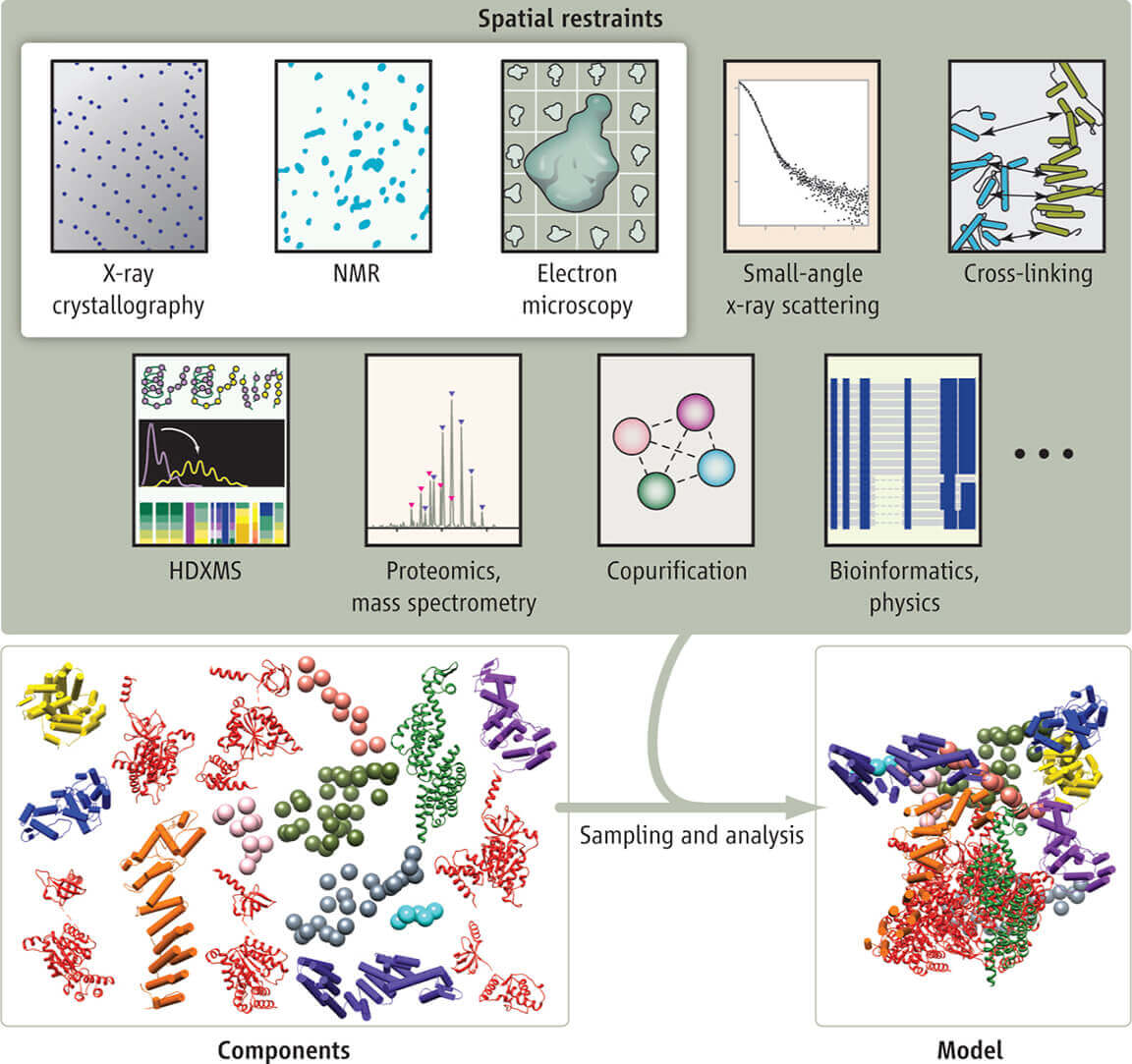
What is Integrative Structural Biology?
Integrative Structural Biology is an interdisciplinary research paradigm that combines various experimental techniques and computational methods to analyze the structure and function of biological macromolecules from multiple scales and dimensions. Its core principle lies in integrating complementary techniques—such as X-ray crystallography, Cryo-EM, NMR, mass spectrometry, and small-angle scattering—to overcome the limitations of individual methods and provide more comprehensive, dynamic structural information.
Importance and Advantages of Integrated Structural Biology Studies
- Complementarity and Precision Enhancement: Each technique has its strengths and weaknesses (e.g., X-ray crystallography offers high resolution but depends on crystals, while Cryo-EM is ideal for large complexes but has lower resolution). When integrated, these techniques can validate and complement each other, offering more precise and reliable results. For instance, Cryo-EM tomography (Cryo-ET) can resolve fine structural details of axons in near-physiological conditions, while mass spectrometry can provide insights into dynamic molecular interactions.
- Dynamics and Functional Correlation: By combining time-resolved crystallography, molecular dynamics simulations, and other techniques, integrated structural biology helps uncover protein conformational changes and their functional mechanisms. A prime example is the study of the in situ structure of photosystem II, where femtosecond X-ray pulses and Cryo-EM were used together to capture rapid structural transitions.
- Complex Systems Analysis: Integrated approaches are particularly powerful for studying complex systems such as membrane proteins and large macromolecular assemblies (e.g., transcription complexes) that traditional methods struggle to handle. The resurgence of in vivo crystallography enables the study of micron-sized crystals under physiological conditions, enhancing our understanding of biological macromolecules in their natural environments.
- Medical Applications: In drug development, the integration of computational models (e.g., pharmacophore modeling) with experimental data accelerates target validation and drug optimization, offering new possibilities for therapeutic intervention.
Select Service
Evolution of Structural Biology Techniques
The field of structural biology has evolved significantly, with key developments in experimental techniques and computational tools, shaping how we study macromolecules.
Early Stage (20th Century)
Foundation of X-ray Crystallography
In 1912, Bragg's Law established the basis for X-ray crystallography. In 1953, Watson and Crick, using Rosalind Franklin's X-ray data, discovered the DNA double helix, marking a major milestone in structural biology.
First Protein Structures
In the 1960s, Perutz and Kendrew determined the 3D structures of hemoglobin and myoglobin, demonstrating that proteins have specific conformations essential for their function.
Limitations
X-ray crystallography required high-quality crystals, which made it difficult to study flexible proteins or membrane-bound proteins.
Transitional Phase (Late 20th Century - Early 21st Century)
Rise of NMR
NMR spectroscopy became important for studying proteins in solution, offering dynamic insights but was limited by sensitivity and size restrictions.
Cryo-EM Breakthrough
In the 2000s, Cryo-EM reached resolutions of 3 Å, enabling the study of large macromolecular complexes without crystals, revolutionizing structural biology.
XFEL and Synchrotron Radiation
In the 2010s, X-Ray Free Electron Laser (XFEL) technology enabled time-resolved studies, capturing protein conformational changes in real-time.
Modern Era (21st Century - Present)
Integration and Multi-Modal Approaches
Today, structural biology integrates various techniques such as Cryo-EM, NMR, and mass spectrometry for more comprehensive and accurate insights, supported by databases like PDB and EMDB.
Advances in Mass Spectrometry
Mass spectrometry (MS) now provides detailed information on protein complexes and interactions, complementing other techniques.
Computational Advances
Molecular dynamics and AI tools like AlphaFold now predict protein structures with high accuracy, enhancing experimental methods.
How Cryo-EM and NMR Work Together
Cryo-EM and NMR are two cornerstone techniques in structural biology, each with its unique strengths and limitations. Their integration provides a multi-dimensional perspective for studying the static structures and dynamic behaviors of complex biological macromolecules. Here's an analysis of their technical complementarity, collaborative mechanisms, and some notable case studies.
Technical Comparison and Complementarity Between Cryo-EM and NMR
A. Resolution and Sample Applicability
| Technique | Resolution | Sample Applicability |
|---|---|---|
| Cryo-EM | Near-atomic resolution (3-4 Å) | Ideal for large molecular complexes (>100 kDa), virus particles, and aggregated fibrils. No need for crystallization and requires minimal sample purity. Example: COVID-19 spike protein (3.5 Å), Zika virus (3.8 Å). |
| NMR | 2-3 Å (Solution NMR), Lower for solid-state NMR | Solution NMR is suitable for small molecules (<25 kDa, newer techniques extend to ~100 kDa). Solid-state NMR can resolve local structures of fibrils and membrane proteins, but with lower global resolution. Best for studying small molecules, flexible regions, and dynamic complexes. |
B. Dynamic Information vs. Static Structure
Cryo-EM excels at capturing high-resolution static structures but struggles with flexible or dynamic regions. For example, in alpha-synuclein fibrils, Cryo-EM clearly defined the core (residues 38-95), while NMR was used to characterize the unobserved N-terminal region, confirming it as highly dynamic and disordered.
NMR, on the other hand, provides detailed dynamic information, such as chemical shift perturbation (CSP) and relaxation rates, which reveals ligand binding, conformational changes, and local flexibility. For instance, fluorine-19 NMR (19F NMR) can detect weak conformational states in membrane proteins, complementing the static images captured by Cryo-EM.
C. Technological Synergy Enhances Model Reliability
- Validation and Optimization: Cryo-EM's 3D reconstruction can be optimized with chemical constraints from NMR data, such as NOE distances and hydrogen bonds, improving the atomic model. For example, integrating NMR data into Cryo-EM models has significantly enhanced the precision of large macromolecular complexes, such as the half-megadalton enzyme complex.
- Multi-Scale Modeling: Cryo-EM provides the overall structural framework, while NMR refines local details, such as the protonation states of active sites or the binding modes of cofactors. This multi-scale approach allows for a more accurate and detailed model of complex biological systems.
Collaborative Mechanisms of Cryo-EM and NMR in Structural Biology
Research of Macromolecular Complexes and Dynamic Regulation
Cryo-EM resolves the core β-sheet arrangement in fibrils (e.g., alpha-synuclein), while solid-state NMR identifies flexible regions or the impact of pathological mutations on stability.
Drug Design and Mechanistic Studies
- Target Identification: Cryo-EM rapidly screens for drug-binding sites, such as in ribosome-antibiotic complexes, while NMR quantifies binding affinities and conformational changes, aiding rational drug design.
- Dynamic Inhibition: In the study of Tc-toxin and F-actin ADP-ribosylation, Cryo-EM captures the overall complex conformation, and NMR reveals the local conformational changes induced by NAD+ binding.
Heterogeneity and Conformational Ensemble Analysis
Cryo-EM can separate primary conformations through 3D classification (e.g., ribosome's rotational states), while NMR detects minor conformations, such as transient intermediates in enzymatic cycles. Together, they create a comprehensive energy landscape of the molecule's conformational space.
Case Studies of Cryo-EM and NMR Collaboration
Case Study 1: Alpha-Synuclein Fibrils in Neurodegenerative Diseases
Cryo-EM: Resolved the β-strand arrangement of the core (residues 38-95) and revealed the effect of pathological mutations (e.g., A53T) on the fibril interface.
Solid-State NMR: Discovered the disordered, dynamic N-terminal region (residues 1-37) and provided insights into its interaction with cell membranes.
Significance: The combined techniques offer a dual view of the structural and dynamic features, aiding the design of inhibitors to prevent fibril growth.
Case Study 2: Temperature Sensing Mechanism of Membrane Protein RSMR
19F NMR: Used to label methionine residues (e.g., M385) and detect conformational states (S4 peak), revealing temperature-induced conformational heterogeneity.
Cryo-EM: Verified the major conformations (IFS and OFS states) but missed the low-abundance intermediate states, highlighting the dynamic advantages of NMR.
Significance: This case underscores how NMR can capture conformational diversity that Cryo-EM may overlook.
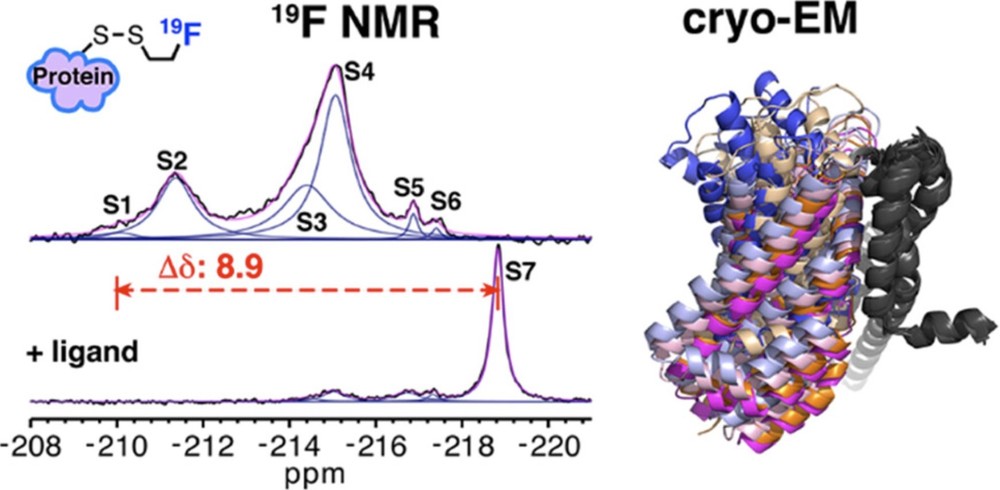 Figure 1. Combining Cryo-EM and 19F NMR for Multistate Conformational Analysis of Membrane Proteins. Using a novel monofluoroethyl 19F probe, changes in membrane protein conformations observed by 19F NMR correlate with cryo-EM structural ensembles. (Huang Y, et al., 2023)
Figure 1. Combining Cryo-EM and 19F NMR for Multistate Conformational Analysis of Membrane Proteins. Using a novel monofluoroethyl 19F probe, changes in membrane protein conformations observed by 19F NMR correlate with cryo-EM structural ensembles. (Huang Y, et al., 2023)
Case Study 3: Atomic Model Optimization of a Half-Megadalton Enzyme Complex
Cryo-EM: Provided the overall structure (4.1 Å).
NMR: Applied constraints on hydrogen bond networks and side-chain orientations, resulting in a high-precision atomic model (RMSD <1 Å).
Significance: This is the first seamless integration of Cryo-EM and NMR data, setting a new standard for studying large complexes.
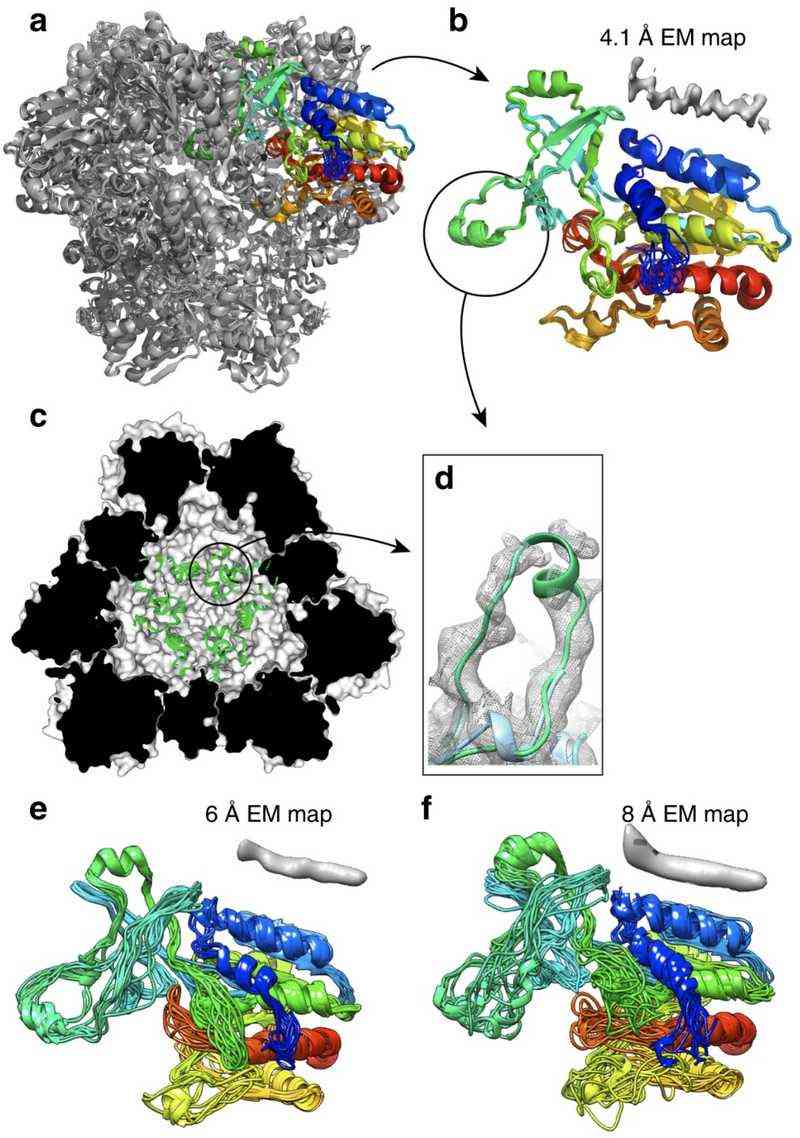 Figure 2. Joint NMR and Cryo-EM Structural Analysis of TET2 Dodecamer. The TET2 dodecamer structure refined using NMR and cryo-EM data, highlighting monomer accuracy and loop region details. (Gauto D F, et al., 2019)
Figure 2. Joint NMR and Cryo-EM Structural Analysis of TET2 Dodecamer. The TET2 dodecamer structure refined using NMR and cryo-EM data, highlighting monomer accuracy and loop region details. (Gauto D F, et al., 2019)
How Cryo-EM and X-ray Crystallography Work Together
Cryo-EM (cryogenic electron microscopy) and X-ray crystallography are two powerful techniques in structural biology that provide detailed insights into the structure and function of biomolecules. They can work together in several ways to complement each other's strengths and overcome their limitations:
A. Complementary Structural Information
Cryo-EM excels at studying large molecular complexes, membrane proteins, and dynamic systems in their near-native states. It can capture multiple conformational states and provide structural information without the need for crystallization.
X-ray Crystallography offers atomic-level resolution and precise details of small molecules and stable protein structures. It is particularly useful for analyzing well-ordered crystals and providing high-resolution insights into binding sites and interactions.
By combining data from both techniques, researchers can obtain a more comprehensive understanding of biomolecular structures. For example, cryo-EM can provide the overall architecture of a large complex, while X-ray crystallography can offer detailed atomic-level information for specific subunits or binding sites.
For a more detailed comparison of cryo-EM and X-ray crystallography, including their complementary roles in structural biology, check out our in-depth guide on Cryo-EM vs. X-ray Crystallography.
B. Joint Analysis in Drug Discovery
Cryo-EM is valuable for visualizing drug targets in their native environment and capturing conformational changes induced by ligand binding. This is especially useful for challenging targets like membrane proteins and large complexes.
X-ray Crystallography provides ultra-high-resolution details of ligand-binding pockets, enabling precise structure-based drug design. The combination of both techniques allows researchers to study drug-target interactions at different scales and states, enhancing the efficiency of drug development.
C. Ligand Identification and Validation
Recent studies have shown that deep learning models trained on X-ray data can be applied to cryo-EM maps to identify ligands. This approach leverages the well-established X-ray datasets and the emerging cryo-EM data to improve the accuracy of ligand recognition.
Combining data from both techniques can also help validate the structural models and improve the reliability of ligand identification.
D. Overcoming Limitations
Sample Preparation: Cryo-EM requires minimal sample purification and can work with smaller sample quantities, while X-ray crystallography demands highly pure and well-crystallized samples. Combining both methods allows researchers to choose the most suitable approach based on sample availability and stability.
Resolution and Computational Needs: X-ray crystallography typically achieves higher resolution for small molecules, while cryo-EM can handle larger complexes and dynamic systems. Additionally, cryo-EM requires extensive computational resources for image processing and 3D reconstruction, which can be balanced by the well-established data processing pipelines of X-ray crystallography.
Case Study: Solving the Structure of TLR13-RNA Complex Using Cryo-EM and X-ray Crystallography
A notable example of the synergy between cryo-EM and X-ray crystallography is the structural determination of the Toll-like receptor 13 (TLR13) in complex with its RNA substrate. TLR13 plays a critical role in immune response by recognizing conserved nucleotide sequences in bacterial 23S ribosomal RNA.
Challenge: The Phasing Problem in X-ray Crystallography
The ectodomain of TLR13, in complex with a 13-nucleotide RNA oligomer (ssRNA13), was successfully crystallized, and its X-ray diffraction data extended to a high resolution of 2.3 Å. However, conventional methods for solving the phase problem—including molecular replacement using homologous structures, heavy atom derivatization, and selenium substitution—proved unsuccessful.
Cryo-EM as a Solution
To overcome the phasing challenge, researchers turned to single-particle cryo-EM. They reconstructed the structure of TLR13 bound to a 25-nucleotide RNA oligomer (ssRNA25) at a resolution of 4.8 Å. This medium-resolution cryo-EM map provided critical phase information, which was then used to successfully phase the high-resolution X-ray crystallographic data.
By integrating cryo-EM reconstruction with X-ray crystallographic diffraction data, researchers resolved the TLR13-ssRNA13 complex structure at 2.3 Å resolution. This case highlights how cryo-EM can serve as an essential tool for overcoming phase determination barriers in X-ray crystallography, particularly for challenging macromolecular complexes.
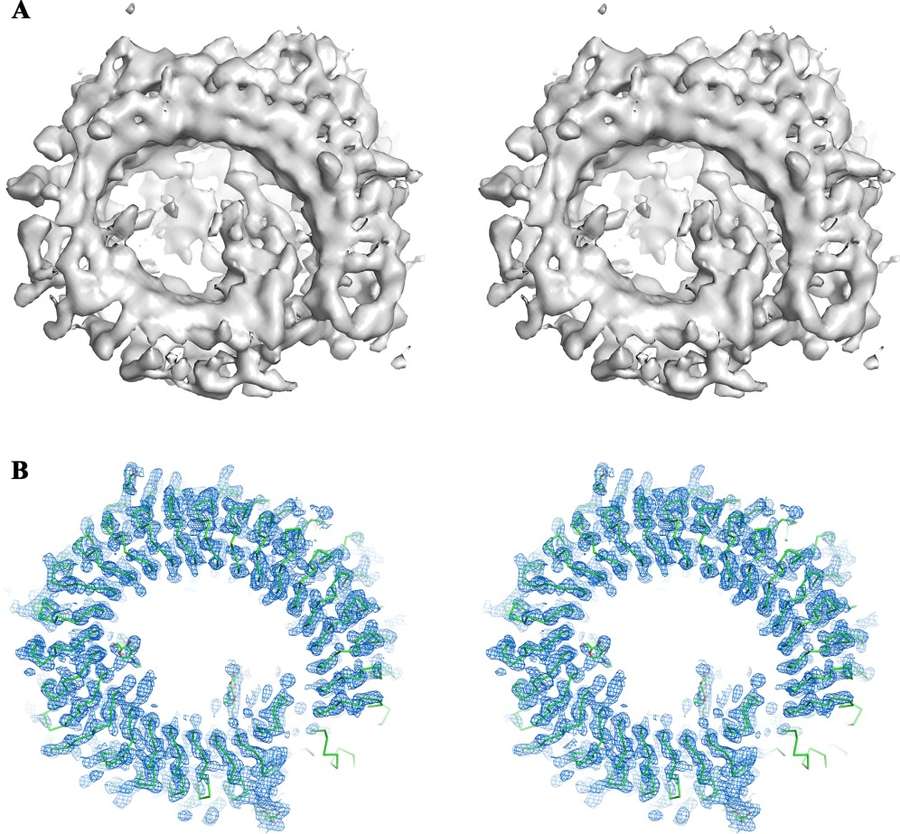 Figure 3. Integration of Cryo-EM and X-ray Crystallography for Structure Determination. (A) Cryo-EM 3D reconstruction of the TLR13-ssRNA complex at 4.8 Å resolution. (B) Electron density map obtained by phase extension from cryo-EM to X-ray crystallographic data at 2.3 Å resolution, showing the atomic model of TLR13. (Xu Y, et al., 2022)
Figure 3. Integration of Cryo-EM and X-ray Crystallography for Structure Determination. (A) Cryo-EM 3D reconstruction of the TLR13-ssRNA complex at 4.8 Å resolution. (B) Electron density map obtained by phase extension from cryo-EM to X-ray crystallographic data at 2.3 Å resolution, showing the atomic model of TLR13. (Xu Y, et al., 2022)
How Cryo-EM and Mass Spectrometry Work Together
Combining Cryo-EM with mass spectrometry (MS) offers powerful new ways to gain deeper and more comprehensive insights into the structures and dynamics of biomolecules. These complementary techniques work synergistically, providing static structural details alongside dynamic, real-time molecular information, which enhances our understanding of complex biological systems.
Complementary Roles of Cryo-EM and Mass Spectrometry
A. Resolution vs. Dynamics
Cryo-EM excels at providing high-resolution static structures of large molecular complexes, however, it is limited when it comes to capturing dynamic changes, such as protein conformational shifts, ligand-binding sites, or post-translational modifications. Mass spectrometry, on the other hand, offers insights into these dynamic processes. Techniques like Hydrogen/Deuterium Exchange Mass Spectrometry (HDX-MS) can track protein conformational changes and identify flexible regions not captured in Cryo-EM.
B. Sample Screening and Validation
Mass spectrometry, especially Native MS, is a powerful tool for rapidly assessing the homogeneity and composition of samples before Cryo-EM analysis. For instance, Native MS can be used to confirm the purity of protein complexes, ensuring that only homogeneous samples are sent for Cryo-EM analysis, thus significantly reducing data collection time and improving the quality of Cryo-EM maps.
C. Comprehensive Analysis of Complex Systems
Cryo-EM may sometimes struggle to resolve the structure of certain complex components, such as ligands or lipids (especially in membrane proteins). In these cases, mass spectrometry can step in to identify the chemical composition of these molecules. For example, lipid mixtures binding to membrane proteins can be identified through mass spectrometry, providing valuable information that complements and refines the Cryo-EM structural model.
Key Mass Spectrometry Techniques Integrated with Cryo-EM
Chemical Cross-Linking Mass Spectrometry (CX-MS)
This technique uses chemical cross-linkers to covalently bond spatially proximal amino acid residues in proteins, which are then identified by mass spectrometry. The cross-linking data provide distance constraints (typically <30 Å) that can be used to model and validate Cryo-EM structures. For example, CX-MS has been used in the structural analysis of the 26S proteasome, where it helped define the interaction network between subunits and aided in the construction of a complete molecular model alongside Cryo-EM data.
Native Mass Spectrometry (Native MS)
Native MS allows for the analysis of intact protein complexes under non-denaturing conditions, providing direct information on their molecular weight, assembly states, and stoichiometry. This technique is particularly useful for sample screening and ensuring uniformity before Cryo-EM analysis. For example, Native MS has been employed in the study of the SARS-CoV-2 replication-transcription complex to confirm its oligomeric state, which ensured the reliability of subsequent Cryo-EM data. Additionally, Native MS can analyze ligand-binding events, offering valuable insights for guiding the focus of Cryo-EM reconstructions.
Hydrogen/Deuterium Exchange Mass Spectrometry (HDX-MS)
HDX-MS monitors the exchange of hydrogen atoms for deuterium, revealing dynamic regions and interaction interfaces on protein surfaces. When integrated with Cryo-EM, HDX-MS can distinguish functionally important areas, such as receptor-binding domains, that undergo dynamic conformational changes. This combination allows for a more comprehensive understanding of static structures and their functional states.
Ion Mobility Spectrometry-Mass Spectrometry (IM-MS)
IM-MS provides insights into the conformational heterogeneity of protein complexes by measuring their shape and size in the gas phase. This technique complements Cryo-EM's sub-classification data, which helps categorize different functional states of a complex. Together, IM-MS and Cryo-EM allow for a deeper analysis of protein complexes with multiple functional states or conformations.
Case Studies of Cryo-EM and Mass Spectrometry Integration
Case Study 1: Structural and Functional Analysis of the SARS-CoV-2 Spike Protein
Cryo-EM Contribution: Cryo-EM was instrumental in revealing the pre-fusion conformation of the SARS-CoV-2 spike protein, specifically capturing the dynamic rotational mechanism of the receptor-binding domain (RBD), which is crucial for the virus's ability to bind to human cells.
Mass Spectrometry Integration:
- Native MS: This technique confirmed the stability of the spike protein trimer, an essential feature for its function as a viral entry protein.
- HDX-MS: This approach identified conformational changes induced by ACE2 binding, shedding light on the differences in affinity between the SARS-CoV-2 and SARS-CoV spike proteins, thus contributing to the understanding of their distinct pathogenicity.
Case Study 2: Integrated Structural Study of the 26S Proteasome
Methodology: The molecular architecture of the proteasome regulatory particle (RP) was reconstructed by combining Cryo-EM with CX-MS and proteomics data.
Findings: This integration revealed the conformational variability of the Rpn1 subunit and the localization of the ubiquitin receptors, Rpn10 and Rpn13. It provided a comprehensive understanding of the synergistic mechanisms involved in substrate recognition and deubiquitination within the proteasome.
 Figure 4. Integrative Structural Analysis of Proteasome Regulatory Particle Using Cryo-EM and Mass Spectrometry. (Lasker K, et al., 2012)
Figure 4. Integrative Structural Analysis of Proteasome Regulatory Particle Using Cryo-EM and Mass Spectrometry. (Lasker K, et al., 2012)
Case Study 3: Cyanobacterial Circadian Rhythm Kai Protein Complex
Mass Spectrometry Role: Native MS was used to monitor the changes in the molar ratios of Kai proteins in real-time, providing crucial information for guiding Cryo-EM experiments to capture the complex at different time points of assembly.
Structural Dynamics: Cryo-EM, in combination with MS, revealed phosphorylation-driven conformational rearrangements within the Kai protein complex, offering insights into the molecular basis of the cyanobacterial circadian rhythm system.
In conclusion, integrative structural biology leverages Cryo-EM, NMR, X-ray crystallography, and mass spectrometry to provide comprehensive molecular insights essential for drug discovery and biomedical research. At Creative Biostructure, we offer expert structural biology services to accelerate your research. Contact us to discuss your project needs!
References
- Lasker K, Förster F, Bohn S, et al. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proceedings of the National Academy of Sciences. 2012, 109(5): 1380-1387.
- Wang H W, Wang J W. How cryo‐electron microscopy and X‐ray crystallography complement each other. Protein Science. 2017, 26(1): 32-39.
- Gauto D F, Estrozi L F, Schwieters C D, et al. Integrated NMR and cryo-EM atomic-resolution structure determination of a half-megadalton enzyme complex. Nature Communications. 2019, 10(1): 2697.
- Olinares P D B, Kang J Y, Llewellyn E, et al. Native mass spectrometry-based screening for optimal sample preparation in single-particle Cryo-EM. Structure. 2021, 29(2): 186-195. e6.
- Huang Y, Reddy K D, Bracken C, et al. Environmentally ultrasensitive fluorine probe to resolve protein conformational ensembles by 19F NMR and Cryo-EM. Journal of the American Chemical Society. 2023, 145(15): 8583-8592.
- Fonda B D, Kato M, Li Y, et al. Cryo-EM and solid state NMR together provide a more comprehensive structural investigation of protein fibrils. Protein Science. 2024, 33(10): e5168.